Amplicon sequencing is known highly targeted gene sequencing method. It is commonly used in analyzing gene variation in specific genomic regions. Amplicons are the products of polymerase chain reaction. Amplicon sequencing is one of the most common NGS-based methods used in target enrichment, along with hybrid capture-based sequencing, which has many applications in clinical and industrial fields. It relies on PCR-amplification of the targeted regions of the genome using sequence-specific primers and probes, and then uses next-generation sequencing techniques. It has two common uses. First, it is used in species identification since diagnostic microbiology utilizes amplicon-based profiling that allows sequencing of regions encoding 16S rRNA. Second, it is also used in the discovery of rare somatic mutations in complex samples. An example of it is tumors mixed with germline deoxyribonucleic acid (DNA).
Next-generation sequencing methods generate high-throughput genomic data. Bioinformatic pipelines, or specific analysis procedures such as amplicon sequencing, are currently developed to be able to extract information of interest from the extremely huge quantity of raw data generated as the product of next-generation sequencing experiments. Since both whole-genome and whole-exome sequencing experiments can only be used in investigating the entire genetic heritage of a person, the targeted re-sequencing methods were introduced for investigating small, user-defined portions of the genome region of interest. These targeted re-sequencing methods are currently widely used in studies involving single- or multi-gene disorders. The commonly adopted targeted re-sequencing solutions are amplicon-based approaches. This kind of approach is based on the synthetic oligonucleotides’ design, also known as probes. Also, the complementary sequence is on the flanking regions of the target genome region to be sequenced. Figure 1 shows an example of the bioinformatics pipeline for next-generation sequencing-based 16S rRNA amplicon sequencing bioinformatics pipeline.
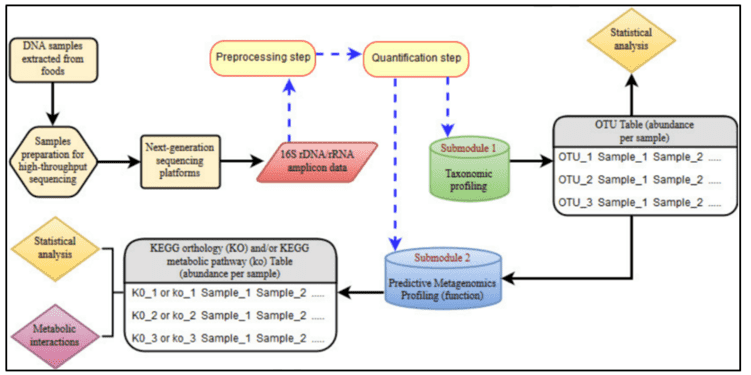 Figure 1. The NGS-based 16S rRNA amplicon sequencing bioinformatic pipeline. (Mataragas, 2018).
Figure 1. The NGS-based 16S rRNA amplicon sequencing bioinformatic pipeline. (Mataragas, 2018).
Amplicon sequencing, also known as ribosomal RNA gene amplification analysis, is an example of the metagenomic/bioinformatic pipeline. The ASAP is an automated way to examine amplicon sequencing data which is also highly customizable. The important details of the amplicon targets include target name, amplicon sequence or the sequences in the case of gene variant assays, regions of interest within the target, and the significance of regions of interest. The mentioned information is described in a text-based input file written in JavaScript Object Notation.
This text-based input file can be hand-generated from an Excel spreadsheet using Phyton Script and a specific provided adapter. The generated sequence is processed by performing adapter and then, quality trimming using Trimmomatic (optional). These sequenced reads are then aligned to the reference amplicon sequences extracted from the JSON file using either of the following alignment packages: (1) bowtie 2, (2) BWA-MEM, and (3) NovoAlign.
The generated BAM file is then analyzed using Pysam with a custom Phython Script, and scikit-bio libraries to aid in the process of analysis. The Python script combines the assay data in the JSON file with the alignment data in the BAM file, and eventually interprets the results. The final output generated is an XML file. This output contains the complete details for each assay against each sample.
References:
Please submit a detailed description of your project. We will provide you with a customized project plan to meet your research requests. You can also send emails directly to for inquiries.
Please fill out the form below: ×