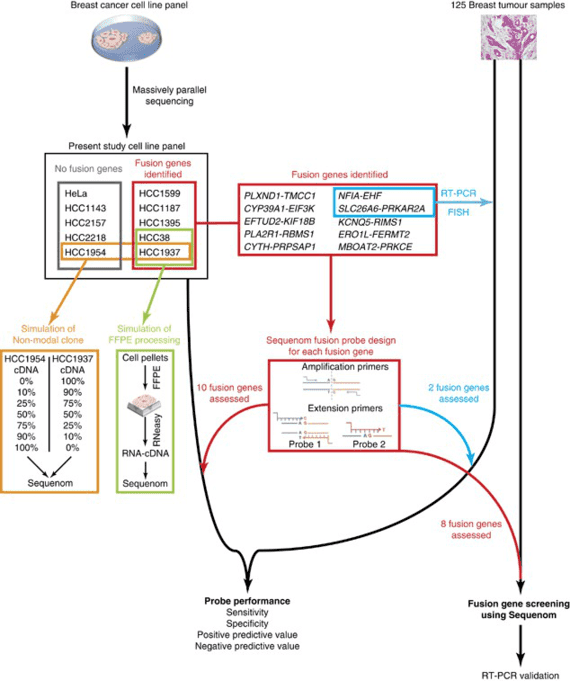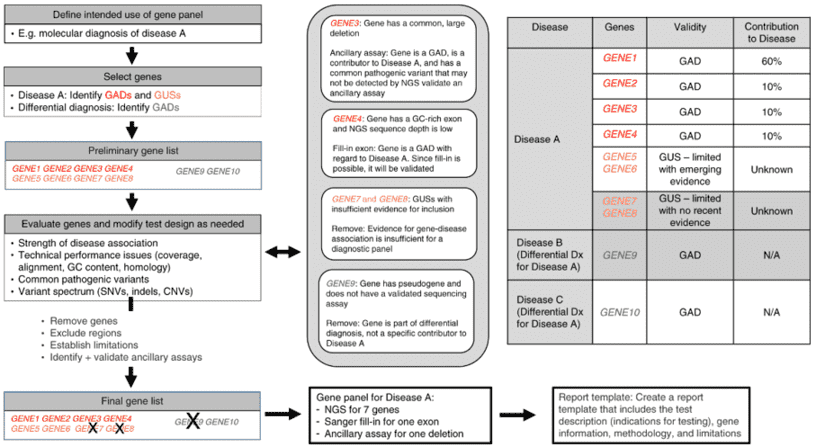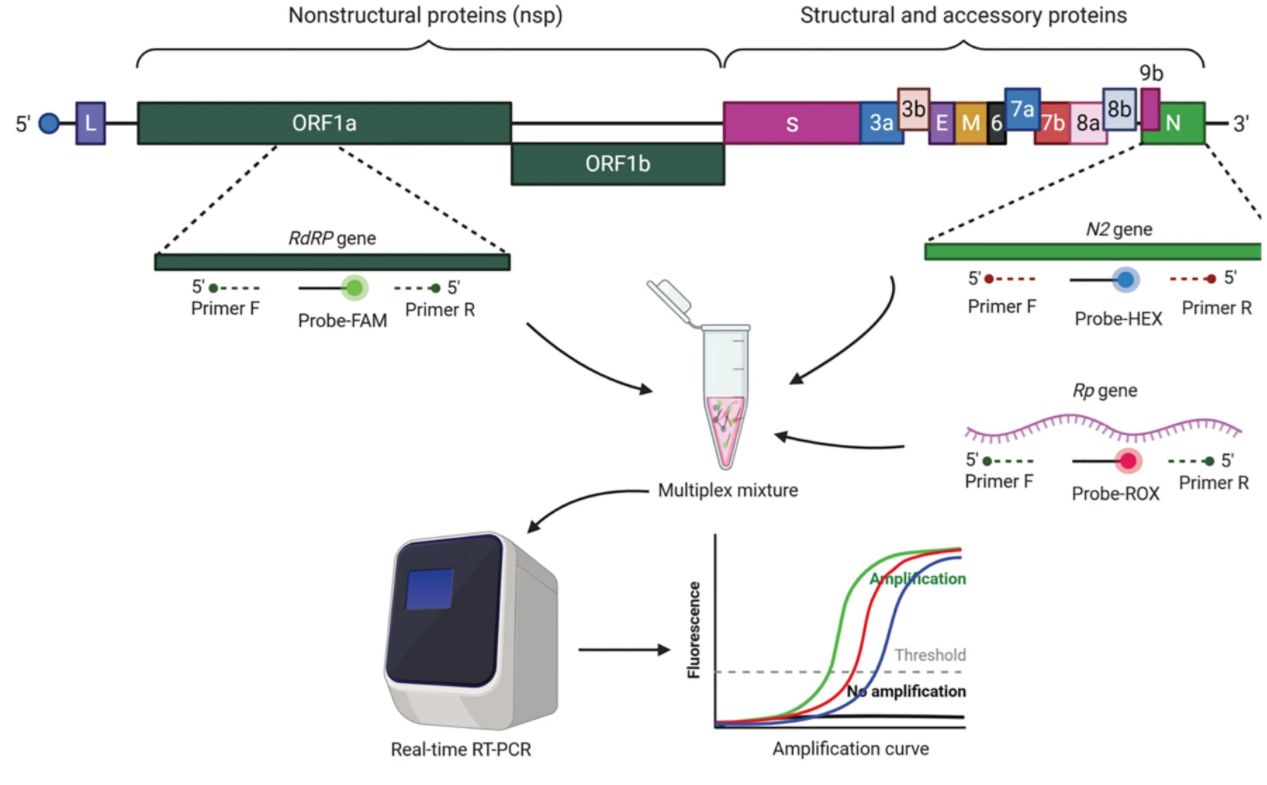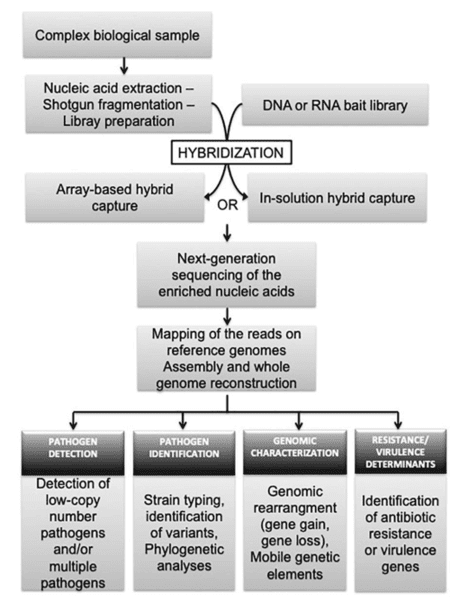
Contracting muscle needs multiple substrate to supply energy. When the process of generating energy in muscle cells is interfered, the cooperation of bones and joints cannot perform properly, causing progressive muscle weakness, fatigue, episodes of pain and cramps after exercise, and/or extensive death and breakdown of muscle tissue, which are some common symptoms of metabolic myopathies. Metabolic myopathies are diseases with defects in enzymes and proteins involved in intermediary metabolism of glucose and fatty acids. It comprises of hereditary (primary) disorders, and acquired (secondary) disorders. Within hereditary disorders, multiple organs are often affected, including respiratory system, etc. Although some people with metabolic myopathies can live normal lives without experiencing significant symptoms due to several pathway to produce ATP, the deficit in ATP can cause severe symptoms when more energy are required to meets body's needs. Three major groups, fatty acid oxidation defects (FAODs), glycogen storage disease (GSD), and mitochondrial myopathies, are mainly inherited.
The mutations of different genes involved in different metabolic progress lead to FAODs, GSD, mitochondrial myopathies and other metabolic myopathies. The deficiency in glycogen metabolism is the main cause of GSD. The McArdle disease (GSD5), the most common metabolic myopathy, is the cause of heterozygous mutations in the PYGM gene, encoding enzyme myophosphorylase. Under normal condition, myophosphorylase initiates glycogen breakdown into glucose-1-phosphate at the early stage of muscle contraction, then gluscose-1-phosphate will be converted into glucose within additional steps. The dysfunction of myophosphorylase will lead to muscle pain and the muscle contracture. Fatty metabolism is also one of the important energy production pathways. 60 mutations in SLC22A5 gene have been reported to cause primary carnitine deficiency, leading to dysfunction of the mitochondrial oxidation of fatty acids. In addition, various mutations in the PNPLA2 gene have been reported in patients with neutral lipid storage diseases associated with myopathy (NLSDM) or neutral lipid storage diseases associated with ichthyosis (NLSDI). More genetic alterations have been found in metabolic myopathies, including PGM1, EPM2A, NDUFS1, ADCK3, TACO1, and so forth.
To have better understanding the mutations in genes involved in metabolic myopathies, our platform provides targeted DNA sequencing by the Illumina MiSeq. You can choose genes of interest for your research from our comprehensive metabolic myopathies panel, covering genes found altered in metabolic myopathies.
| ABHD5 | ACAD9 | ACADM | ACADS | ACADVL |
| ADCK3(CABC1) | BCS1L | C12orf65 | COQ2 | COQ9 |
| DYSF | EARS2 | ECHS1 | ENO3 | EPM2A |
| GBE1 | GFER | GFM1 | GYG1 | GYS1 |
| LAMP2 | LDHA | LIPT1 | LPIN1 | LRPPRC |
| NDUFA1 | NDUFA10 | NDUFA12 | NDUFA2 | NDUFA4 |
| NDUFAF5 | NDUFAF6 | PFKM | PGAM2 | PGK1 |
| PGM1 | PHKA1 | PHKA2 | PYGM | RBCK1 |
| SCO2 | SLC19A3 | SLC22A5 | SLC25A20 | SLC25A3 |
| SLC25A4 | TAZ | TK2 | TEME70 | TMEM126B |
(Download the epilepsy gene list for more genes)
For more information about the Custom Metabolic Myopathies Panel or need other amplification requirements, please contact us.
References:
Please submit a detailed description of your project. We will provide you with a customized project plan to meet your research requests. You can also send emails directly to for inquiries.
Please fill out the form below: ×