Leber congenital amaurosis (LCA) is mostly autosomal recessive, accounting for more than 5% of hereditary retinal diseases. LCA is the most severe hereditary retinopathy that can cause a baby to lose vision completely within one month of birth. In addition to causing congenital blindness in infants, LCA is accompanied by a series of serious complications such as neurological deafness, obesity, diabetes, diabetes insipidus, hypogonadism, hyperuricemia and hypertriglyceridemia. Early fundus examinations may be no abnormalities or mild hyperpigmentation, with search-like nystagmus and pupillary light-reflecting, showing a black-sexual pupil or a quenching electroretinogram waveform. In the late stage, the salt-like pigment and bone cell-like pigmentation are observed, and the electroretinogram shows no symptoms such as no waveform or severely reduced amplitude. The cause of this disease is unknown and there is no effective treatment.
In recent years, many pathogenic genes associated with LCA have been discovered, and there is a considerable gap in the frequency of distribution of these genes among different populations. Among them, AIPL1, CEP290, CRB1, GUCY2D and RPE65 are the most common pathogenic genes, accounting for 50% to 60% of all LCA cases. AIPL1 is involved in the development of cones and rod cells. Therefore, mutations in this gene may cause abnormalities in the transport of retinal-related proteins, thereby affecting the normal function of photoreceptor cells. Mutations in the CEP290 gene are often detected in LCA patients, and intron mutations in CEP290 will result in abnormal exons in CEP290 mRNA. The biochemical function of CRB1 is currently unclear. Comparison of CRB1 with homologous fruit fly dander (CRB) suggests that this protein may be involved in the development of retinal neurons. Therefore, mutations in this gene may lead to retinal dysfunction and thus correlate with LCA. Mutations in the P858S and L954P sites in the GUCY2D gene significantly attenuate the effects of cGMP production and guanylate cyclase activating protein, resulting in a continuous low level of intracellular cGMP, a gated ion channel that is continuously shut down, and a decrease in intracellular Ca2+ concentration. The sensor cells are hyperpolarized for a long time, which is equivalent to their long-term exposure to light, causing degeneration and dysfunction. Mutations in the RPE65 gene result in a decrease in rhodopsin content of the retina, which is equivalent to a long-term dark environment in the retina. In recent years, the study has discovered the new pathogenic gene NMNAT1 of LCA. The mechanism by which the NMNAT1 mutant gene causes LCA is not clear, but most studies attribute the cause of the disease to a decrease in protease activity caused by the NMNAT1 mutation.
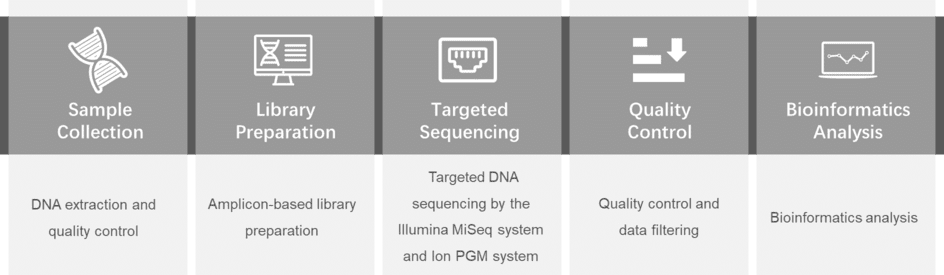
To better understand and support for the study of genetic alterations in Leber congenital amaurosis, CD-Genomics provides a custom Leber congenital amaurosis panel containing optimized genes which are reported associated with the increase of risk of Leber congenital amaurosis. Targeted DNA sequencing technology by Illumina MiSeq/Ion PGM system is provided to detect the genes you are interested in.
| AIPL1 | CABP4 | CEP290 |
| CRB1 | CRX | DTHD1 |
| GDF6 | GUCY2D | IFT140 |
| IMPDH1 | IQCB1 | KCNJ13 |
| LCA5 | LRAT | NMNAT1 |
| OTX2 | PRPH2 | RD3 |
| RDH12 | RPE65 | RPGRIP1 |
| SPATA7 | TULP1 |
For more information about the Custom Leber Congenital Amaurosis Panel or need other amplification requirements, please contact us.
References:
Please submit a detailed description of your project. We will provide you with a customized project plan to meet your research requests. You can also send emails directly to for inquiries.
Please fill out the form below: ×