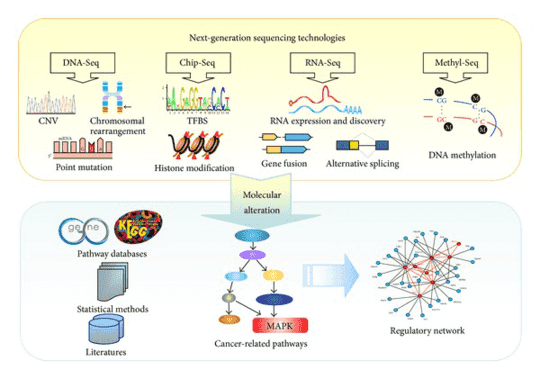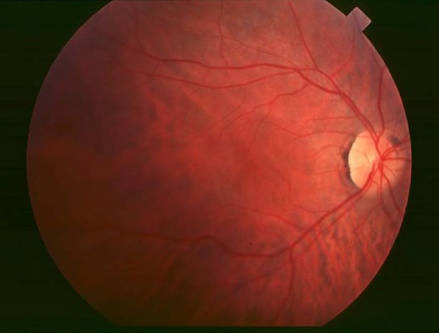
Retinitis pigmentosa (RP) is described as a kind of retinal degeneration disease which often causes progressive loss of photoreceptors. This disease represents a retinal dystrophy with an extremely complex pathogenesis, and the symptoms are further worsened with the impairment of the retinal vascular supply. The prevalence of retinitis pigmentosa is approximately about 1:4000. It is often associated with the night blindness and eventually blindness after several decades. Up to date, there are numerous genetic mutations been identified as the causes of RP, which can explain the heterogeneity of the disease. MERTK gene mutation is a common cause of RP.
MERTK, located on chr2q13, is responsible for the efficient phagocytosis of shed photoreceptor outer segments (POS) by the retinal pigment epithelium. Mutations in the ERTK gene disrupt phagocytosis, leading to the accumulation of exfoliative POS and the formation of subretinal debris, which results in the formation of RP. The mutations of MERTK gene account for approximately 1% to 2.5% in all the RP cases. PRPF31 is mapped on chr19q13.42. The mutations in PRPF31 can cause retinitis pigmentosa by haploinsufficiency. PRPF31 is found to account for 2.5% of autosomal dominant retinitis pigmentosa. Most PRPF31 pathogenic mutations are single-base changes or deletions that leads to premature termination of codons and nonsense mediated mRNA decay. Otherwise, PRPF31, PDE6 (PDE6A, PDE6B, PDE6G), RP25 and RPE65 also have higher prevalence in RP cases.
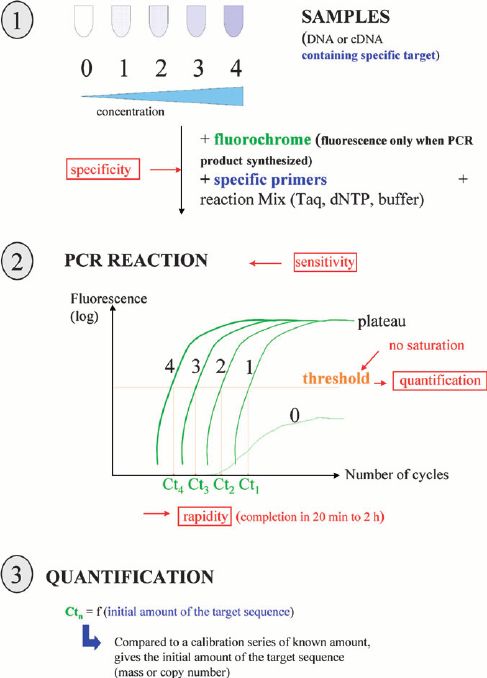
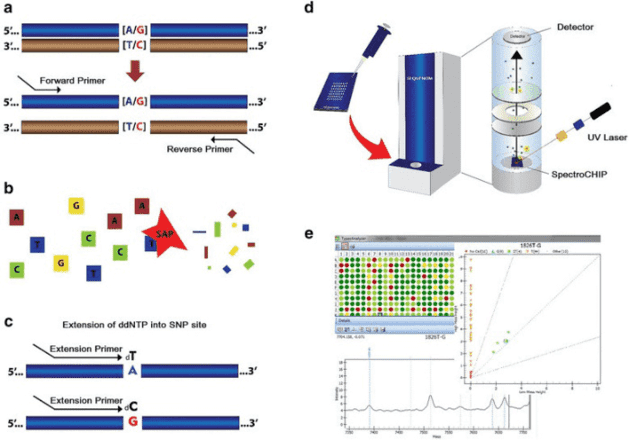
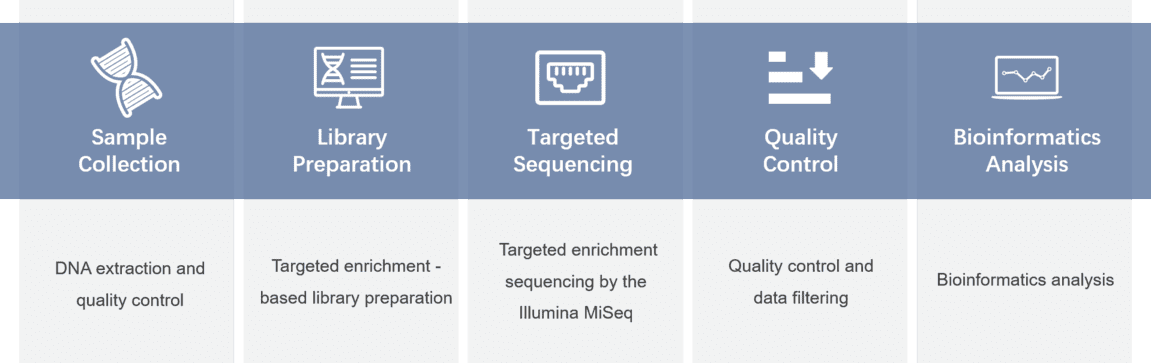
To better understand the consequences of these mutations of MERTK and other associated genes, our platform provides targeted DNA sequencing by the Illumina MiSeq or Ion PGM system, and offers a comprehensive retinitis pigmentosa panel library from which you can choose for genetic testing of retinitis pigmentosa.
| ABCA4 | ABHD12 | AGBL5 | AIPL1 | ARHGEF18 | ARL6 |
| BBS2 | BEST1 | C2ORF71 | C8ORF37 | CA4 | RCDHR1 |
| CERKL | CLRN1 | CNGA1 | CNGB1 | CRB1 | CWC27 |
| CYP4V2 | DHDDS | DHX38 | FAM161A | FLVCR1 | IDH3B |
| IMPG2 | KIZ | KLHL7 | LRAT | MAK | MERTK |
| MFRP | NR2E3 | NRL | PDE6A | PDE6B | PDE6G |
| PEX7 | PHYH | PRCD | PRPF | RBP4 | RDH12 |
| RDH5 | REEP6 | RGR | RHO | ROM | RP1 |
| RP2 | RPE65 | RPGR | RS1 | SAG | SAMD11 |
| SEMA4A | SLC7A14 | SNRNP200 | SPATA7 | SPP2 | TTC8 |
| TULP1 | TOPORS | USH2A | WDR19 | ZNF408 | ZNF513 |
For more information about the Custom Retinitis Pigmentosa Panel or need other amplification requirements, please contact us.
References:
Please submit a detailed description of your project. We will provide you with a customized project plan to meet your research requests. You can also send emails directly to for inquiries.
Please fill out the form below: ×