
The choice of approaches available depends on the priorities of sequencing and requires precision, quality, and cost tradeoffs. Most clinical and public health microbiology laboratories use short-read sequencing systems for routine sequencing, which create sequence fragments up to 1000 base-pair long. While microbial genomes are usually smaller and less complex than human genomes, techniques of long-read sequencing such as real-time single-molecule sequencing are helpful for creating complete, highly precise genomes and figuring out plasmids, repeats, and other complex areas.
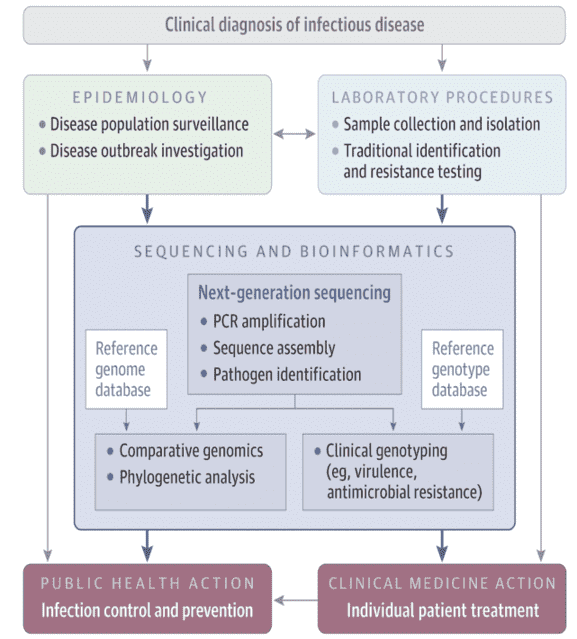 Figure 1. Workflow Converting genome sequence data from pathogens into credible data. (Gwinn, 2019)
Figure 1. Workflow Converting genome sequence data from pathogens into credible data. (Gwinn, 2019)
The translation of raw sequence data into actionable data is dynamic and computer-intensive (Figure 1). Usually, the first step is to compile shorter fragments into a complete sequence, either by mapping against a known reference genome or by using overlapping reads to arrange the sequence de novo. Many separate generalizations, such as pathogen recognition, high-resolution strain typing, and prediction of major phenotypic characteristics, are enabled by comparing the constructed genome with reference strains. Well set and updated reference libraries are vital because microbial pathogens are rapidly emerging and bacteria can share plasmids through strains and organisms, also encoding virulence and antimicrobial resistance characteristics. As proof of replication, integrated genomes may be contrasted with others to search for phylogenetic clustering. Each phase involves various bioinformatics methods that must be harmonized into a coherent workflow: assembling, strain typing, phenotyping, and clustering.
In public health, sequencing of the next decade provides important benefits in terms of speed and resolution of sequence variations for monitoring and disease investigation. For example, in PulseNet, a foodborne disease surveillance device, the transition to the next sequencing from an older molecular approach is in progress. Now, PulseNet is able to identify outbreaks faster, more reliably discern clusters of similar events, and more easily connect pathogens to possible polluted food supplies. The convergence of epidemiology with pathogen genomics is improving public health efforts to prevent the dissemination of communicable diseases such as tuberculosis. Isolates of genotyping tuberculosis may corroborate transmission inferred from touch inquiries or indicate ties between seemingly different events, helping health departments prioritize their efforts better. Next-generation sequencing has the ability to provide more rapid information on possible sensitivity to anti-mycobacterial drugs than traditional research, allowing for more precise and timely treatment.
Next-generation sequencing knowledge is capable of standardization and exchange, a significant benefit for public health collaboration such as the influenza monitoring scheme of the World Health Organization. A transparent approach to "sequence first" will help to generate timely and reliable results to the collection of candidate influenza vaccines, detecting predominant variants rapidly while tracking the complexities of co-circulating viral populations. For difficult field investigations, next-generation sequencing often provides benefits. For instance, during the 2015 Ebola outbreak, a research team from the United Kingdom bundled a nanopore sequencing laboratory into normal luggage for transport to Guinea. 142 Ebola virus genomes were sequenced on-site over an 8-month period normally within 1 working day; information was sent to the cloud for processing and findings were returned the next day. The team provided actionable disease response information without transporting samples from the region amid major logistical difficulties, including inadequate electrical power and internet access.
References:

