
Next-generation sequencing has been a great help for understanding the characteristics of cancer, as well as for potential therapeutic approaches for cancer for the past several years. Aside from its use in identifying novel cancer mutations, NGS is also utilizing in genetic testing of hereditary cancer syndrome, detecting circulating cancer DNA, and designing personalized cancer treatment. With the great help of NGS technologies, base-pair level sequencing of the entire genome can be accomplished with minimum errors, and at the same time, at a lower cost. One of the most powerful tools for cancer research that utilizes NGS technologies is called the cancer panel, also known as a gene panel, a test used in analyzing multiple genes at once for cancer-associated mutations.
Genome testing modality is the beginning of the clinical workflow for the analysis, as well as interpretation of the cancer genome. This ends with the classification of the clinical effect of the variant and reporting of the results which is relevant to the oncologists.

Quality Assessment
The first step in the analysis and interpretation of the cancer genome is the quality assessment of the NGS reads. In this step, the quality NGS reads is evaluated to either remorse, correct, or trim those that did not meet the standards. Also, errors such as base-calling errors, as well as poor quality reads are assessed.
Aligning Sequences
After the assessment of quality, the reads are then aligned to the reference genome. Some of the most common sources of the human reference genome are the University of Santa Cruz (UCSC), and Genome Reference Consortium (GRC). In selecting alignment, two common issues that may arise are the following: (1) solving the problem of ambiguity in mapping short reads to the reference genome, and (2) mismatched reads caused by mutations must be discarded before undergoing further analysis.
Identifying Variants
This very important step in the data analysis has the sequence as the main parameter. The tools used for variant identification are divided into four categories: (1) germline callers, (2) somatic callers, (3) copy number variants or CNV identification, and lastly (4) structural variants or SV identification.
Annotating Variants
This step provides biological significance as it identifies disease-causing variants. The annotation of single nucleotide polymorphisms (SNPs), and insertions/deletions (INDELS) is done by using computational annotation tools.
Visualization of Next-Generation Sequencing
Using visualization tools and genome browsers, the NGS data are visualized after the process of annotations. Through this step, information about variants such as mapping quality, aligned reads, and annotation information like a consequence, scores of different annotation tools, and impact variants are obtained.
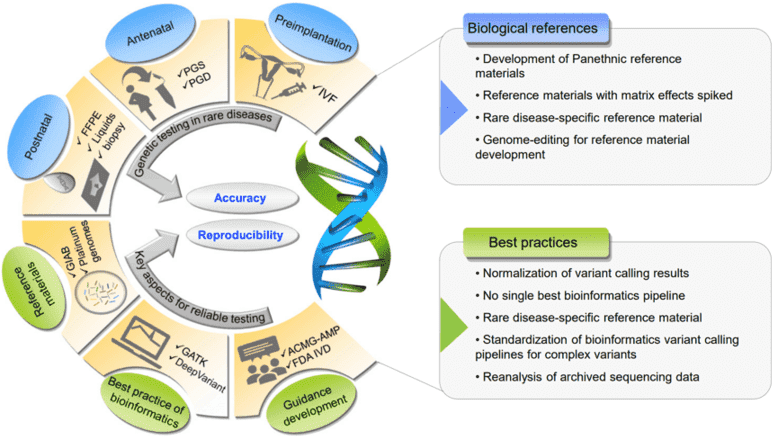 Figure 1. The outstanding challenges of cancer panels and potential solutions for enhancing accuracy and reproducibility. (Liu, 2019)
Figure 1. The outstanding challenges of cancer panels and potential solutions for enhancing accuracy and reproducibility. (Liu, 2019)
NGS-based cancer sequencing has been used for cancer diagnosis. Many commercial, as well as academic laboratories, uses such diagnostics as they have the capacity to identify a full coverage of hereditary variation. This offers the possibility to streamline testing through the utilization of a single analysis platform. Single-cell sequencing is usually used for the characterization of cancer heterogeneity, which is caused by different factors such as rare cells, tissue hierarchies, dynamic cell states, and clonal evolution. Results can be used for clinical purposes such as prognosis and treatment.
References:

