The ability of next-generation sequencing (NGS) to easily produce vast amounts of sequencing data per cycle, with the associated reduction in sequencing costs has redefined human clinical study. It is now possible to obtain unbiased ultra-deep sequencing of complex samples, while bioinformatics analyses can still be lengthy and repetitive. The particular issue in the field of infectious disease detection where the rapid recognition and functional classification of a certain pathogen is crucial for the clinical treatment of infected patients. The gold standard approach for the clinical diagnosis of infectious diseases as of now is polymerase chain reaction (PCR). This approach can give information on the presence/absence and abundance of a targeted microbial pathogen, and is focused on the amplification of a generally short and conserved genomic region. PCR is known to have several benefits, but microorganisms with an extremely diverse sequence cannot be identified because of PCR's high specificity. Also, only partial data on genetic variation, genotype, functional ability, dietary needs, and virulence or antibiotic resistance can be given by PCR.
In these situations, the influence of NGS may also be of particular significance in the reconstruction of complete genomes of pathogens directly from nucleic acids isolated from clinical samples. However, NGS can fail to detect/reconstruct genomes from pathogens present in low quantities in the sample due to the low pathogen/nucleic acid ratio. Capture methods such as hybridization capture accompanied by NGS sequencing (also referred to as hybrid-capture sequencing or target-enrichment sequencing) are applied directly to human clinical specimens that have been created to address these limitations. Hybrid capture enables large genomic components to be collected to complete high-sequencing genomes, enabling downstream research such as phylogenetics, ecology, epidemiology, and drug tolerance.
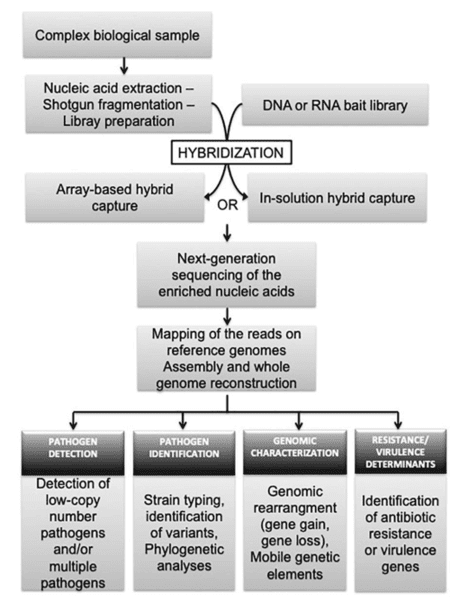 Figure 1. Overview of target target-enrichment sequencing procedure and its application to research and diagnostic infectiology. (Gaudin, 2018)
Figure 1. Overview of target target-enrichment sequencing procedure and its application to research and diagnostic infectiology. (Gaudin, 2018)
Next-generation sequencing hybridization-based capture is a method that is specifically implemented after isolation of nucleic acid and preparation of the library. Fragmented shotgun libraries are denatured by heating and exposed to single-stranded oligonucleotide hybridization with DNA or RNA (also called 'probes' or 'baits') unique to the area of interest. RNA baits are preferable since, relative to DNA: DNA hybrids, RNA: DNA duplexes are stronger in terms of hybridization success and stability.
Probe design is a key step in the study of target enrichment (TES) since it influences the performance of hybridization of probes and the efficacy of TES. The design of the probe also establishes what genomic regions for enrichment and sequencing to target. It is possible to design probes from transcribed sequences.
The unbound non-specific molecules are drained out and for NGS, the enriched DNA is eluted. It is possible to perform hybridization between DNA libraries and baits in a solution or on decent support. DNA probes are linked to rigid support in the solid process, such as a glass microarray slide, where free DNA or RNA probes are biotinylated in the solution-capture, enabling them to separate heteroduplexes of the desired fragment-probe using magnetic streptavidin beads. There is no systematic technique for target enrichment procedures so far and several changes can be made to the size of the library fragment, sample fragmentation, cleanup procedures, number of cycles of PCR amplification, and/or the length of hybridization. The last approach usually involves trimming steps (for adapter sequences, poor quality, and redundant reads), mapping the remaining reads for pathogen detection and recognition of reference genomes, and/or assembling them into contiguous genome reconstruction (Figure 1). Sequencing the enhanced nucleic acids and bioinformatic analyses of the reads are the final stage in NGS hybridization-based capture.
References:
Please submit a detailed description of your project. We will provide you with a customized project plan to meet your research requests. You can also send emails directly to for inquiries.
Please fill out the form below: ×