The function of urea circulation is to synthesize toxic ammonia into non-toxic urea, so as to avoid the occurrence of hyperammonemia. The cycle includes a series of six steps of biochemical reactions. There are 6 kinds of enzymes involved in the urea cycle: N-acetylglutamate synthase (NAGS), carbamoyl phosphate synthetase I (CPSI), ornithine transcarboxylase (OTC), argininosuccinate synthetase (ASS), argininosuccinate lyase (ASL) and argininase (ARG). In order to maintain the normal urea cycle, aspartic acid, the reaction substrate of AS, must be transported from mitochondria to cytoplasm through citrin, the carrier of mitochondrial intima, while ornithine, the reaction substrate of OTC, must be transported from cytoplasm to mitochondria through ORNT1, another transporter of mitochondrial intima. The occurrence of structural or functional defects in the above 6 enzymes, 2 carriers or any of the transporters will affect the synthesis of urea and form hyperammonemia which is urea circulation disorder (UCDs).
In genetics, except for OTC deficiency which is linked to X chromosome, all the other 5 enzymes are autosomal recessive. Mutations in the genes encoding NAGS, CPSI, OTC, ASS, ASL and ARG enzymes will result in dysfunction of the above enzymes, obstruction of urea circulation, inability of ammonia to form non-toxic urea, and then, ammonia can't be excreted. In addition to the pathogenic mutations of the above enzymes, there are other gene mutations associated with other UCD types. Mutations in the SLC25A15 gene on chromosome 13q14 result in hyperornithinemia-hyperammonemia-homocitrullinuria (HHH) syndrome. The SLC25A15 gene encodes the transporter ORNT1 which is involved in the urea cycle. The mutation in SLC25A13 gene leads to citrin vector defect, which cannot transport aspartic acid from mitochondria to the cytoplasm to participate in urea cycle. The mutations in SLC7A7 gene can lead to defective y (+) LAT-1 protein which expresses in the basilateral membrane of small intestine and renal tubular epithelial cells, and result in lysinuric protein intolerance. Mutation of the GLUD1 gene causes a defect in its encoded glutamate dehydrogenase (GDH), a decrease in sensitivity to the allosteric inhibitor GTP, and an increased sensitivity to the allosteric activator leucine, resulting in sustained GDH activity and hyperinsulinism/hyperammonemia (HI/HA).
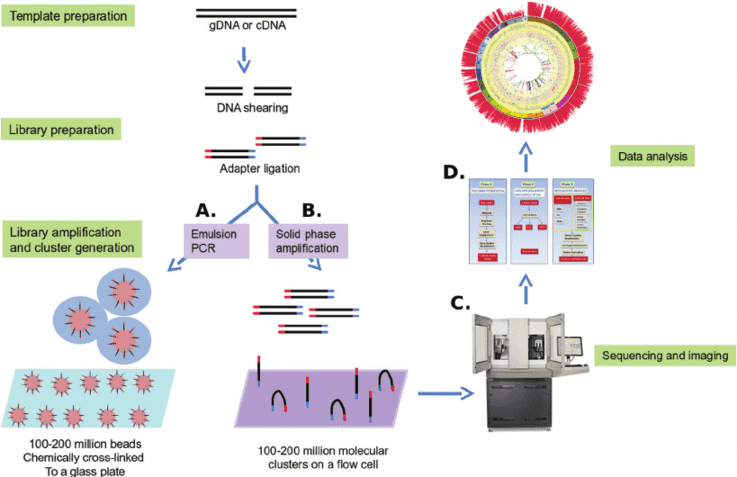
To better understand and support for the study of genetic alterations in urea cycle disorder, CD-Genomics provides a custom urea cycle disorder panel containing optimized genes which are reported associated with the increase of risk of urea cycle disorder. Targeted DNA sequencing technology by Illumina MiSeq system is provided to detect the genes you are interested in.
| ACADM | ACADS | ACADVL | ALDH18A1 | ARG1 | ASL |
| ASS1 | ATP5F1E | ATPAF2 | BCKDHA | BCKDHB | BCS1L |
| BTD | CA5A | CPS1 | CPT1A | CPT2 | DBT |
| DLD | ETFA | ETFB | ETFDH | GLUD1 | HADHA |
| HADHB | HCFC1 | HLCS | HMGCL | HMGCS2 | IVD |
| MCCC1 | MCCC2 | MMAA | MMAB | MMACHC | MMADHC |
| MUT | NAGS | OAT | OTC | PC | PCCA |
| PCCB | PYY | PIGA | SERAC1 | SLC25A13 | SLC25A15 |
| SLC7A7 | SUCLA2 | SUCLG1 | TMEM70 | TTC19 | TUFM |
| UQCRB | UQCRQ |
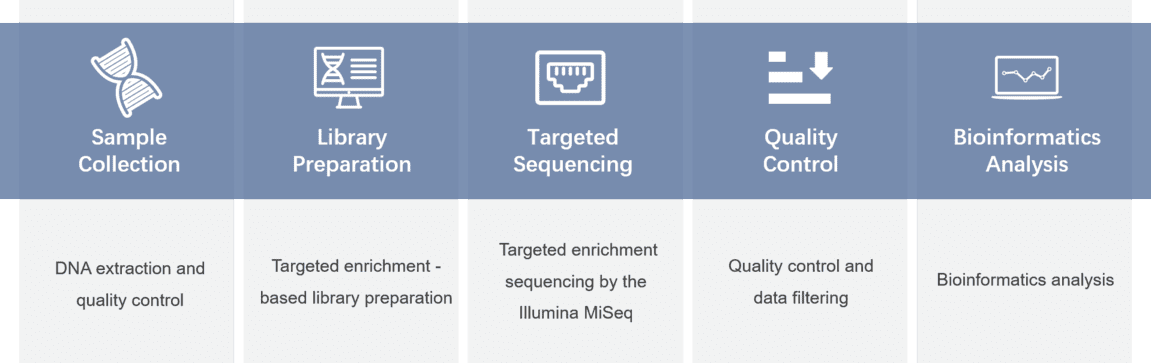
For more information about the Custom Urea Cycle Disorder Panel or need other amplification requirements, please contact us.
References:
Please submit a detailed description of your project. We will provide you with a customized project plan to meet your research requests. You can also send emails directly to for inquiries.
Please fill out the form below: ×