Next-generation sequencing (NGS) has sparked an uprising in disease panel. NGS involves libraries to be prepared in which DNA or RNA molecules (fragments) are bonded with adapters, preceded by PCR amplification and sequencing. Robust methods of library preparation that generate a representative, non-biased source of nucleic acid material from the genome under investigation are of crucial importance. The first phase of next-generation sequencing is library preparation. It enables the adherence of DNA or RNA to the sequencing flow cell and enables the identification of the sample. Two common library preparation methods are library prep based on ligation and library prep based on tagmentation.
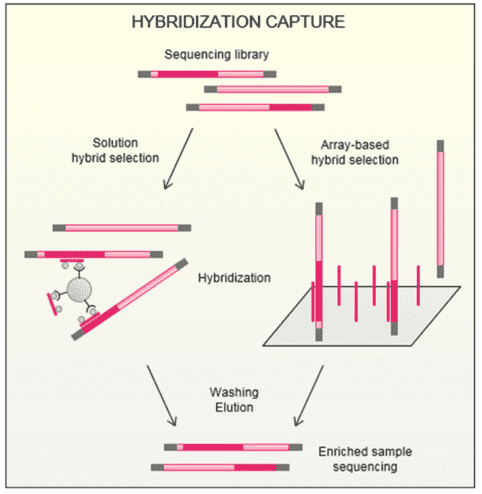 Figure 1. Principle of hybridization capture. (Gasc, 2016)
Figure 1. Principle of hybridization capture. (Gasc, 2016)
DNA or RNA samples must be fragmented, end-repaired, and collected into adapter-ligated libraries before they can be sequenced by next-generation sequencing. The result obtained by NGS experiments can be influenced by library preparation protocols. In general, the key steps in preparing for NGS analysis of RNA or DNA are: (i) fragmenting and/or sizing the target sequences to the desired length, (ii) converting the target to double-stranded DNA, and (iii) attaching adapters of oligonucleotides to the ends of the target fragments.
Fragmenting and/or sizing the target sequences to the desired length
An important factor for NGS library construction is the size of the target DNA fragments in the final library. To fragment nucleic acid chains, there are three techniques accessible: physical, enzymatic, and chemical. Usually, the fragmentation of DNA is performed by physical techniques. Digestion by DNase I or Fragmentase includes enzymatic methods. Both were found to be successful in comparing NGS libraries built with acoustic shearing/sonication versus fragmentase. However, relative to the physical processes, Fragmentase provided a higher number of artifactual indels.
Converting target to double-stranded DNA
The RNA would be fragmented in most cases before translation into cDNA. This is normally achieved with a divalent metal cation (magnesium or zinc) by the use of controlled heated digestion of the RNA. By increasing or minimizing the time of the digestion reaction with strong reproducibility, the optimal duration of the library insert can be modified.
Attaching oligonucleotide adapters to the ends of target fragments
To optimize the library size and eliminate adaptor dimers or other library planning artifacts, a second post-library design sizing step is widely used. Adapter dimers are the product of the adapters' self-ligation without a series of library inserts. These dimers very effectively form clusters and use precious space without producing any useful data on the flow cell. In most cases, the first operates for samples where the adequate starting material is available. More adapter dimer products are frequently created when sample input is reduced.
Bias
When planning a sequencing library, the primary aim is to establish as little bias as possible. Due to experimental design, bias can be defined as the systematic manipulation of results. Since all causes of experimental bias can not be removed, the strongest methods are: (i) understanding where bias exists and taking all possible steps to mitigate it and (ii) paying attention to experimental design so that the sources of bias that can not be eliminated have a small effect on the final result.
Complexity
An NGS library's complexity can represent the amount of bias produced by a given experimental design. The ideal, in terms of library complexity, is a highly complex library that represents the original complexity of the source material with high fidelity. The technical difficulty is that this fidelity can be reduced by some amount of amplification. The complexity of libraries can be calculated by the amount or percentage of repeat reads contained in the sequencing results. Duplicate reads are commonly described as reads that when aligned to a reference sequence, are completely similar or have the precise same start positions. One caveat is that with the sequencing depth, the number of repeat reads that arise by chance (and reflect genuinely separate sampling from the original sample source) increases. It is also necessary to consider under what circumstances duplicate read rates constitute a specific measure of library complexity.
Complexity
An NGS library's complexity can represent the amount of bias produced by a given experimental design. The ideal, in terms of library complexity, is a highly complex library that represents the original complexity of the source material with high fidelity. The technical difficulty is that this fidelity can be reduced by some amount of amplification. The complexity of libraries can be calculated by the amount or percentage of repeat reads contained in the sequencing results. Duplicate reads are commonly described as reads that when aligned to a reference sequence, are completely similar or have the precise same start positions. One caveat is that with the sequencing depth, the number of repeat reads that arise by chance (and reflect genuinely separate sampling from the original sample source) increases. It is also necessary to consider under what circumstances duplicate read rates constitute a specific measure of library complexity.
Batch effect
It is also important to take into account the reduction of batch effects when planning libraries for NGS sequencing. It is also necessary to consider the influence of systemic bias arising from the molecular manipulations needed to produce NGS data; for instance, the bias added in miRNA-seq library preparations by sequence-dependent variations in adaptor ligation efficiencies. Differentiation in day-to-day sample preparation, such as reaction conditions, reagent batches, the precision of pipetting, and even separate technicians, can lead to batch effects. In addition, during sequencing runs and between separate lanes on an Illumina flow-cell, batch effects can be observed. It may be quite straightforward or very complicated to minimize batch effects. During the experimental design method, contacting a statistician when in question will save an enormous amount of lost resources and time.
References:
Please submit a detailed description of your project. We will provide you with a customized project plan to meet your research requests. You can also send emails directly to for inquiries.
Please fill out the form below: ×