Custom Spinal Muscular Atrophy Panel
What is spinal muscular atrophy?
Spinal muscular atrophy (SMA) is one of the most common fatal neuromuscular diseases due to degeneration of motor neurons in the anterior horn of the spinal cord, resulting in proximal muscle symmetry, progressive atrophy and weakness, ultimately leading to respiratory failure and even death. It is the second fatal autosomal genetic disease which belongs to autosomal recessive genetic disease.
SMA is clinically heterogeneous. Patients are classically divided into one of the five types according to their age of onset and peak motor function achieved. Type 0 is the most severe form of the disease. It mainly occurs during pregnancy, reducing fetal movement and development, resulting in respiratory distress at birth. Type I, also known as Werdnig Hoffmann's disease, occurs before 6 months of age and has significant muscle atrophy and inability to sit up by themselves. The profound hypotonia can manifest as a "frogleg" posture when lying, and be poor to have head control. They have a life expectancy of less than three years. Type II disease occurs mainly between 6 and 18 months. Many of the comorbidities in this patient population are related to the orthopedic complications of bone and joint development in the setting of muscular weakness and progressive scoliosis, and joint contractures and ankylosis of the mandible may develop. Life expectancy of type II patients is between 10 and 40 years. Type III, also known as Kugelberg-Welander disease, occurs after 18 months of age and may result in losing walking ability in adulthood. The mildest type of SMA, named type IV, produces muscle weakness in adulthood, but the individual maintains walking ability. Types III and IV usually do not reduce life expectancy.
Disease-related gene description
SMA has a broad range of onset age, severity, progression rate and variability between subtypes. 95% of cases of SMA, irrespective of type, are caused by a homozygous deletion in the
SMN1 gene on chromosome 5q13. SMN protein is ubiquitously expressed and is a part of multiprotein complexes that probably have both general and motor neuron specific functions. There are two forms of the
SMN gene exist on each allele: a telomeric form (SMN1) and a centromeric form (
SMN2).
SMN2 is a homologous gene of
SMN1. It has 5 different nucleotides compared to latter. The 6th nucleotide of exon 7 of SMN2 gene is mutated from cytosine (C) to thymine (T), which leads to the change of
SMN2 pre-messenger RNA cleavage site and makes the deletion disappear of exon 7, and then expresses a truncated SMN protein that is defective in function and rapidly degraded. Variation in
SMN2 copy numbers, which partly explains differences in SMN protein levels between patients, is the most important modifier of SMA severity.
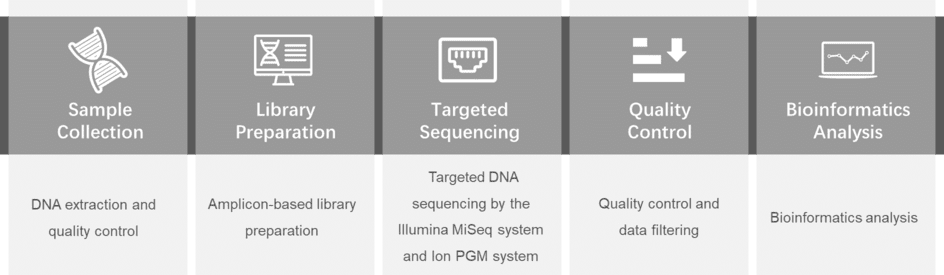
Targeted DNA sequencing with the Illumina MiSeq or Ion PGM system makes it an ideal platform for fast and cost-effective genetic analysis in a wide range of applications. CD Genomics offers an abundant spinal muscular atrophy panel library, from which you can choose what you want for your researches.
Custom spinal muscular atrophy panel offers but are not limited to:
- Provide related gene information consultation and custom panel services to save your cost and increase efficiency.
- Utilize the highly multiplexed and polymerase chain reaction (PCR) based workflow by the Illumina MiSeq to provide a highly targeted sequencing, which is developed as a quick, accurate and cost-effective method to identify genetic mutations associated with spinal muscular atrophy.
- The results of detected genetic variant will be further validated to verify the sequencing accuracy.
- Custom panel genes are constantly updated based on the frontiers of literature studies to target all relevant regions.
Choose the genes that suit you from the spinal muscular atrophy gene list
| AARS |
ASAH1 |
ATP7A |
BICD2 |
| BSCL2 |
CHCHD10 |
DCTN1 |
DNAJB2 |
| DYNC1H1 |
EXOSC3 |
EXOSC8 |
FBXO38 |
| GARS |
HEXA |
HSPB1 |
HSPB3 |
| HSPB8 |
IGHMBP2 |
LAS1L |
PLEKHG5 |
| REEP1 |
SCO2 |
SLC5A7 |
SMN1 |
| SMN2 |
TBCE |
TRPV4 |
UBA1 |
| VAPB |
VRK1 |
|
|
Specimen requirements of our custom spinal muscular atrophy panel
- Specimen: whole blood, saliva or extracted DNA (not FFPE-compatible).
- Volume: min. 1ml blood; min. 2 μg extracted DNA.
- Collection: blood and saliva are collected in EDTA tube. DNA samples are stored in TE buffer or equivalent.
Gene panel workflow
For more information about the Custom Spinal Muscular Atrophy Panel or need other amplification requirements, please contact us.
References:
- Farrar MA, et al. Emerging therapies and challenges in spinal muscular atrophy. Annals of Neurology, 2017, 81(3):355-368.
- Lefebvre S, et al. Identification and characterization of a spinal muscular atrophy-determining gene. Cell, 1995, 80(1):155-65.
- Kolb SJ, Kissel JT. Spinal Muscular Atrophy. Neurologic Clinics. 2015, 33(4):831-46.
- Nash LA, et al. Spinal Muscular Atrophy: More than a Disease of Motor Neurons? Current Molecular Medicine, 2016, 16(9):779-792.
- Simone C, et al. Is spinal muscular atrophy a disease of the motor neurons only: pathogenesis and therapeutic implications? Cellular and Molecular Life Sciences, 2016, 73(5):1003-20.
* For research purposes only, not intended for clinical diagnosis, treatment, or individual health assessments.
Related Services
Related Products
Related Resources