Spinocerebellar ataxia (SCA) is a genetic disorder characterized by slow progressive gait. It is often associated with poor coordination of hand, speech and eye movements. Spinocerebellar ataxia causes great inconvenience to the life of patients and their families. Spinocerebellar ataxia has many types, and each of them could be considered a neurological condition in its own right. This disease is caused by the mutation of either a recessive or dominant gene. SCA can affect anyone of any age, and is currently considered to be a progressive and irreversible disease. There is no cure for SCA till now. The treatment now we use are directly towards alleviating symptoms, but can’t cure the spinocerebellar ataxia. The study of spinocerebellar ataxia-related genes is an important way to help us explore the pathogenesis of SCA and develop new therapies.
Although there are many types of SCA, there are not many related genes. Till now we find 5 genes related, they are ATXN1, ATXN2, ATXN3, ATXN6, ATXN7, respectively. The different loss-of-function mutations in ATXN1 have distinct phenotypes, which explains why researchers only find five strong-related genes but SCA has many types. Protein encoded by ATXN1 forms a transcriptional repressor complex with Capicua, and the complex leads to neurodegeneration which makes SCA happens. The mutations of CAG repeat expansions in the middle sequence of ATXN2 and ATXN3 can also increase the risk of SCA. Most of the mutations in SCA-related genes cause the repeat expansions of trinucleotide. These expansions occur between two generations of unaffected individuals and promote the emergence of an ataxia phenotype. But ATXN7 appears to cause SCA in a different way. Mutations in ATXN7 cause the expansion of the polyglutamine tract and lead to the development of debilitating neurodegenerative diseases.
CD-Genomics provides a custom SCA panel which contains optimized genes related to SCA. You can customize the gene panel depend on your own requirements. Targeted sequencing technology powered by Illumina MiSeq system/Ion PGM system ensures to investigate the SCA related genetic variations discovered in routine clinical practice.
| ATXN1 | ATXN2 |
| ATXN3 | ATXN6 |
| ATXN7 |
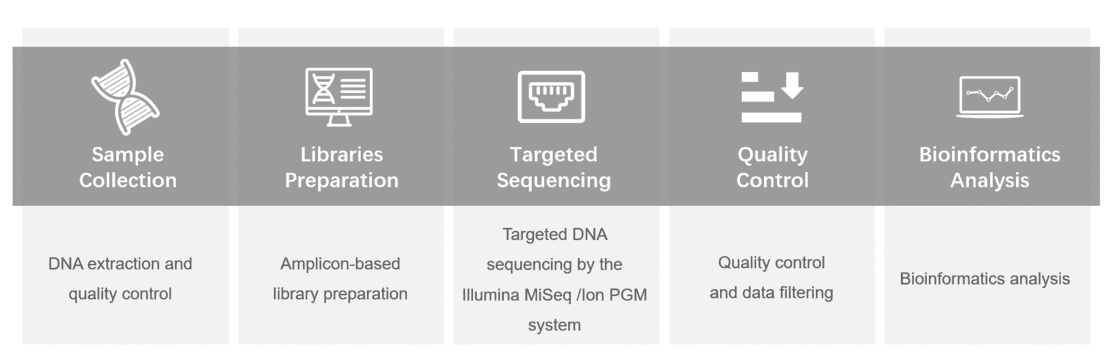
For more information about the Custom SCA Panel or need other amplification requirements, please contact us.
References:
Please submit a detailed description of your project. We will provide you with a customized project plan to meet your research requests. You can also send emails directly to for inquiries.
Please fill out the form below: ×