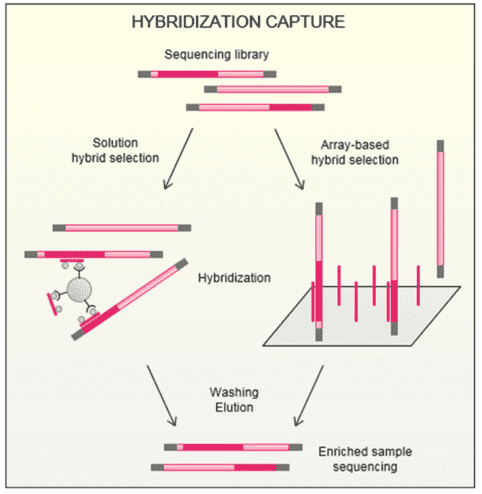
Congenital myasthenic syndrome (CMS) is a group of conditions characterized by muscle weakness that worsens with physical exertion. Symptom of muscle weakness usually begins in early childhood but can also happens on adult. CMS usually affects facial muscles, like the muscles that control the eyelids and move the eyes, and the muscles used for chewing and swallowing. In fact, any of the muscles used for movement can be affected in this condition. Affected infants may have feeding difficulties due to muscle weakness. Congenital myasthenic syndrome always gets worse triggered by fever or infection. Fortunately, CMS is curable and the incidence is low. Studying the genes related to CMS can accelerate your disease mechanism research, biomarker discovery, and targeted drug development.
Congenital myasthenic syndromes are rare and caused by mutations in genes that encode proteins necessary for neuromuscular transmission. It is well known that CHAT encodes the choline O-acetyltransferase. The mutations in CHAT can affect the conversion of choline and acetyl-CoA to acetylcholine subsequently, causing the greatly reduced release of acetylcholine to the neuromuscular junction, and resulting in the failure of appropriate neuromuscular transmission. GFPT1 is involved in the synthesis of UDP-GlcNAc, which is a nucleotide sugar used as a building block for glycosylation pathways of acetylcholine. The mutations in GFPT1 cause the abnormal glycosylation of acetylcholine receptor subunits, leading to the failure of neuromuscular transmission. COLQ encodes the triple-stranded collagenic tail thats include an N-terminal proline-rich attachment domain. ColQ anchors in the synaptic space, extend the lifespan of Ach in synapse space. Mutations of COLQ cause the nerve terminals to be abnormally small and curtail the number of Ach available for release, which leads to CMS. SLC25A1 is a mitochondrial citrate carrier which mediates the exchange of citrate/isocitrate with cytosolic malate. Mutations in the SLC25A1 are associated with severe neurometabolic disease. Research has found three unrelated CMS families carrying the same missense mutations in SLC25A1, presenting with a similar phenotype, confirming the association between genotype and phenotype.
| AGRN | ALG14 | ALG2 |
| CHAT | CHRNA1 | CHRNB1 |
| CHRND | CHRNE | COLQ |
| DOK7 | DPAGT1 | GFPT1 |
| GMPPB | LAMB2 | LRP4 |
| MUSK | PLEC | PREPL |
| RAPSN | SCN4A | SLC5A7 |
| SNAP25 | SLC25A1 |
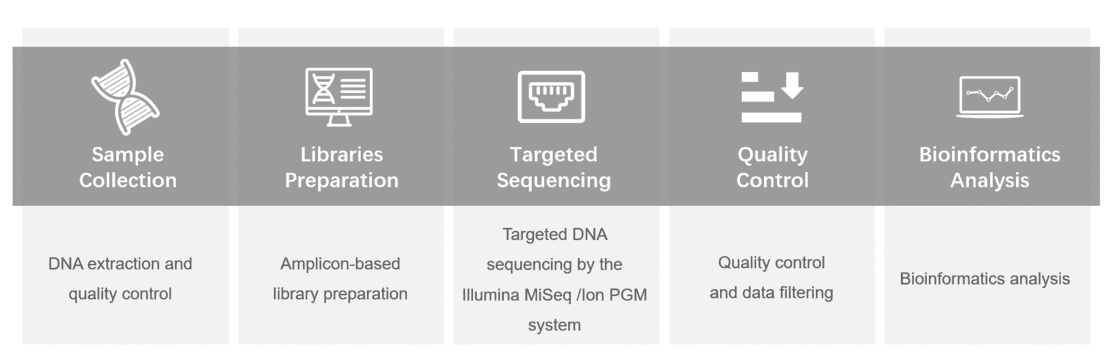
For more information about the Custom Congenital Myasthenic Syndrome Panel or need other amplification requirements, please contact us.
References:
Please submit a detailed description of your project. We will provide you with a customized project plan to meet your research requests. You can also send emails directly to for inquiries.
Please fill out the form below: ×