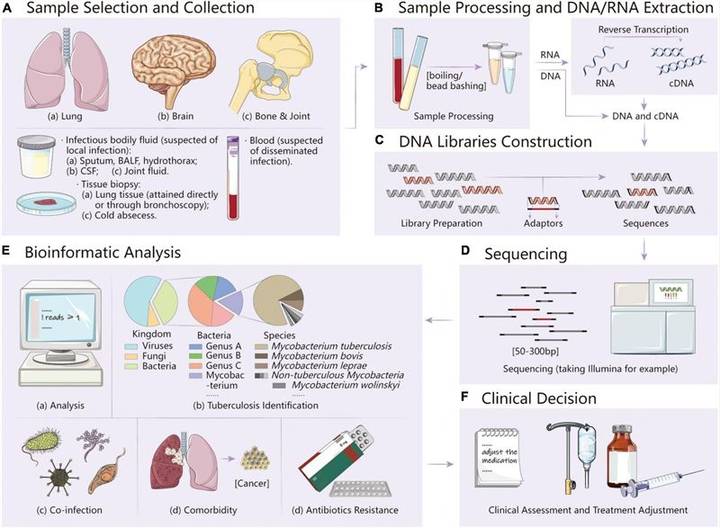
Chronic Granulomatous Disease (CGD) is an inherited immune system disorder with malfunction of phagocyte (neutrophils, monocytes, marcrophages and eosinophils) resulting from impaired killing of bacteria and fungi. In most situations, people with CGD have at least one serious bacterial or fungal infection every 3 to 4 years. Patients with CGD usually develop pneumonia, lymphadenitis, abscess, osteomyelitis, abscesses or cellulitis. Cluster of white blood cells may gather in the infectious areas, forming granulomas. It is estimated that one individual suffers from CGD in 200,000 to 250,000 people worldwide. Most of the people are diagnosed before the age of five, but CGD may present at any time from infant to adulthood. Generally, patients with X-linked CGD which is the altered gene located on the X chromosome, have a more severe disease syndrome with earlier age and show and death.
The bacterial and/or fungal infection will stimulate phagocyte and activate nicotinamide adenine dinucleotide phosphate (NAPDH) oxidase. Upon activation, phosphorylated p47phox can interact with p22phox after conformational changes, which leads to membrane translocation of p47phox with activation of GTPase-Rac and forms the active oxidase complex by assembling p40phox and p67phox. The cooperation of ROS and proteins from granules mediates the killing of the pathogens. The defects in the NAPDH oxidase complex will cause CGD. The autosomal recessive CGD (AR-CGD) is caused by mutations in CYBA (encoding p22phox), NCF1 (encoding p47phox), NCF2 (encoding p67phox) and NCF4 (encoding p40phox); the X-linked recessive CGD (XR-CGD) results from mutations in the CYBB gene (encoding gp91phox). The most common mutations in CYBB will lead to different variants: X910 (protein and oxidase activity absent), X91− (low levels of mutated protein and residual oxidase activity) and X91+ (mutated protein normally expressed but oxidase activity absent). What’s more, the mutations in AR-CGD are related to activity of NADPH oxidase in A470 (protein absent), A220/+ (mutated protein absent or normally expressed) and A67−/0 (low level or absent mutated protein). Besides, two variants of Nucleotide-binding oligomerization domain-containing protein-2 (NOD2), IVS8(+158) and R703C are found in CGD patients. Recently, deficiency of glucose-6-phosphate dehydrogenase (G6PD) and Ras-related C3 botulinum toxin substrate 2 (Rac2) genes have also been reported to lead to insufficient NADPH formation in leucocytes and insufficient signal transduction from surface receptors to the NADPH oxidase, respectively.
To have better understanding of these mutations of genes related to CGD, targeted DNA sequencing by the Illumina MiSeq or Ion PGM system will be provided by our platform. This panel includes 7 genes associated with CGD and you can choose from these 7 genes or have your own customized panel.
| CYBA | CYBB |
| Rac2 | G6PD |
| NCF1 | NCF2 |
| NCF4 | NOD2 |
For more information about the Custom Chronic Granulomatous Disease Panel or need other amplification requirements, please contact us.
References:
Please submit a detailed description of your project. We will provide you with a customized project plan to meet your research requests. You can also send emails directly to for inquiries.
Please fill out the form below: ×