The DNA segments that do not encode protein sequences are known as non-coding DNA sequences. It represents more than 98% of the human genome sequence. They had previously been called “junk DNA”, but then eventually rehabilitated by the Encyclopedia of DNA Elements project as researchers uncovered that approximately 80% of the human genome is functional. Several discoveries, such as the enrichment location of single nucleotide polymorphisms (SNPs) is within or near non-coding functional elements, emphasized the huge potential of investigating variants in medical research.
Non-coding elements include cis-regulatory regions such as promoters, enhancers, insulators, and silencers, as well as non-coding RNAs. Most of these elements were able to identify using the combination of functional genomic approaches and in-silico methods based on sequence conservation. These segments exhibit histone modification patterns which is a characteristic of specific functional elements.
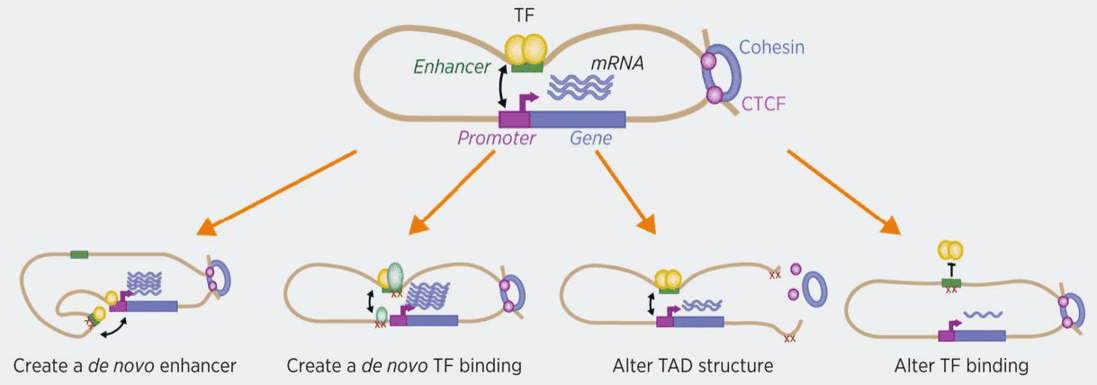 Figure.1 Potential mechanisms of action of noncoding mutations.
Figure.1 Potential mechanisms of action of noncoding mutations.
Types of non-coding sequences were established based on their functions. The first type of non-coding sequences is the regulatory elements that control gene expression. These specifically include cis-regulatory elements which are the sequences located in 5’ or 3’ untranslated regions (UTRs) or within introns. As mentioned, these cis-regulatory elements may act as enhancers, promoters, or silencers of gene expression when bound by trans-regulatory elements, such as TFs or transcription factors. There are transcribed sequences that are not translated into peptide products that can also be involved in gene expression regulation. These transcribed sequences include introns, as well as several classes of non-coding RNAs such as microRNA (miRNA), transfer RNA (tRNA), and long non-coding RNA (lncRNA).
Non-coding variants can be disrupted either by single nucleotide variants or SNVs, insertions, and deletions of less than 50 base pairs or indels, or larger structural variants (SVs). The mentioned variants can be found at the germline level which means either they can be inherited and be found in all cells of an individual, or they can appear in somatic cells at any point in the lifetime of an individual. Such mutations can cause diverse consequences depending on the specific location of the disrupted sequence. Several somatic non-coding mutations have been discovered to be associated with cancer. One of the most powerful technologies that could target specific genes of mutation that are associated with cancer is by using cancer panels that utilize next-generation sequencing approach.
First, the gain of transcription factors-binding site (TF-binding site) which is exemplified with the promoter of TERT or the telomerase reverse transcriptase, the catalytic subunit of telomerase. There are studies conducted recently which reported the recurrent mutations in the TERT promoter in different cancer types.
Second, the fusion of active regulatory elements in oncogenes, genes associated with cancer, because of structural rearrangements. Such phenomenon frequently happens in 5’ UTR of TMPRss2 and ETS family genes in prostate cancer. This fusion results in the overexpression of ERG, which then inhibits the signaling of the androgen receptor and subsequently malignant transformation in the prostate. Aside from that, it has been found out that modifications in ncRNAs or their binding sites can be a signature of cancer since lncRNA MALATI was observed to regulate the expression of genes that are associated with metastasis and upregulated in lung cancer.
Finally, transcribed pseudogenes are discovered to affect, as well as regulate their parental counterparts. This has been documented using miRNA sequestration for the BRAF gene which is a gene associated with several cancer types such as breast cancer.
References:
Please submit a detailed description of your project. We will provide you with a customized project plan to meet your research requests. You can also send emails directly to for inquiries.
Please fill out the form below: ×