
Wheat Genomes Analysis: Insights into Sequencing, Assembly, Annotation, and Evolution
Wheat feeds about one third of the world's population, and its yield and quality are directly related to global food security. However, the wheat genome is huge and complex, with highly repetitive sequences, and the polyploid characteristics make its genome sequencing and assembly a very challenging task. With the continuous development of sequencing technology, a series of breakthrough progress has been made in wheat genome sequencing, which has laid a solid foundation for further understanding the biological characteristics of wheat and accelerating genetic improvement.
This paper introduces many analysis methods in wheat genome research, and emphasizes its important significance for crop improvement and breeding paradigm transformation.
Wheat Genome Sequencing and Assembly
Wheat is an important food crop in the world, its genome sequencing and assembly is of great significance for analyzing complex biological characteristics and promoting the process of genetic breeding.
In 2005, the International Wheat Genome Sequencing Alliance (IWGSC) was established, which started the journey of wheat genome sequencing. Early research mainly focused on sequencing a single chromosome of wheat, such as the wheat 3B chromosome sequencing project, and gradually analyzed the chromosome sequence through BAC-by-BAC (bacterial artificial chromosome) strategy. These studies have accumulated valuable experience for the follow-up whole genome sequencing, but limited by technology and cost, the progress is relatively slow.
With the progress of sequencing technology, the whole genome sequencing of wheat ushered in a breakthrough. In 2018, IWGSC released the reference sequence of Zhongchun wheat genome, which is an important milestone in wheat genome research. By integrating various sequencing techniques and assembly strategies, the genome has been assembled at chromosome level, covering most areas of the wheat genome. Since then, the genomes of more wheat varieties have been sequenced and assembled, such as the high-quality genome sequence of bread wheat variety "Fielder", which further enriched the wheat genome resources.
In order to improve the quality of genome, researchers constantly optimize and update the published genome. By increasing the sequencing depth, introducing new technical means, filling gaps, correcting errors, improving the accuracy of gene annotation, making the wheat genome sequence more complete and accurate, and providing a more reliable data basis for in-depth study of gene function and regulation mechanism.
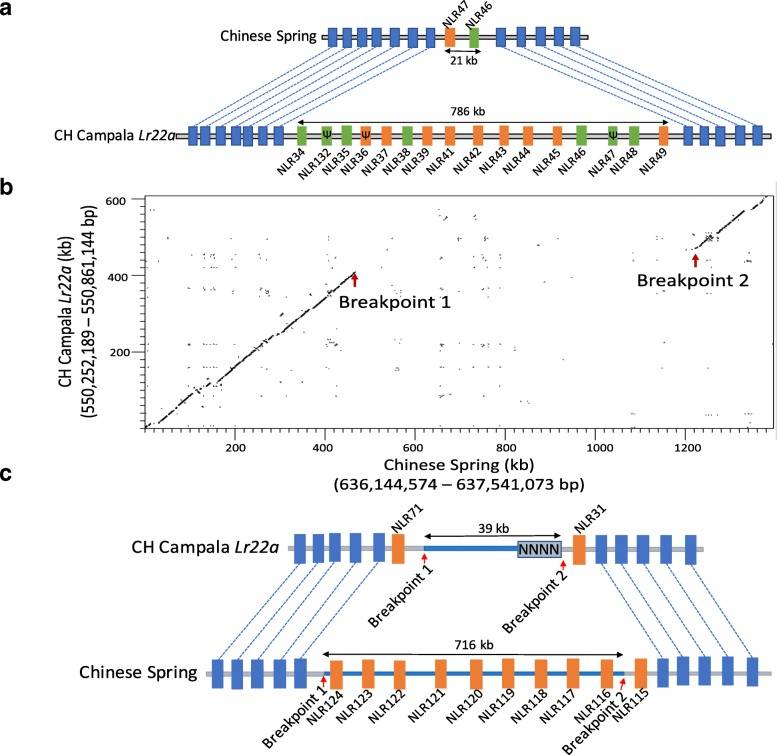
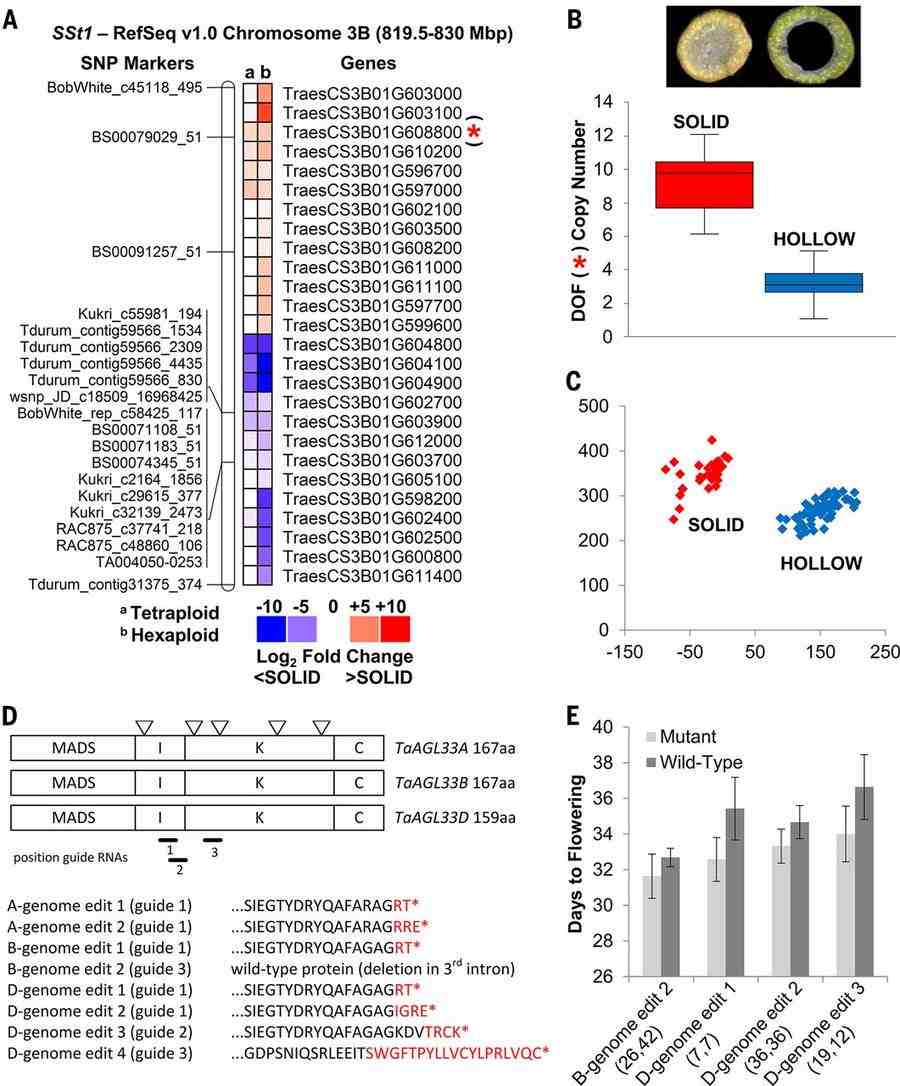 IWGSC RefSeq v1.0–guided dissection of SSt1 and TaAGL33 (IWGSC et al., 2018)
IWGSC RefSeq v1.0–guided dissection of SSt1 and TaAGL33 (IWGSC et al., 2018)
Analysis of Wheat Genome Complex Regions
The complex region of wheat genome contains many key genes, which are closely related to the growth and development, environmental adaptability, yield and quality of wheat. Therefore, it is of great significance to deeply analyze the complex regions of wheat genome for analyzing the biological characteristics of wheat and cultivating new varieties with high yield, high quality and stress resistance.
Characteristics of Complex Regions of Genome
Repeat sequence: The proportion of repeat sequence in wheat genome is very high, accounting for 85%-90%, in which transposon is the main component. Long terminal repeat (LTR) retrotransposons are widely distributed in the genome, and their active transpositions not only increase the size of the genome, but also affect gene expression through insertion or regulation.
Polyploid characteristics: The allohexaploid origin of wheat makes its genome contain subgenomes (A, B, D) from three different ancestors. There are extensive homology and gene redundancy among subgenomes, but some genes have been lost, sequence differentiation and expression patterns have changed during evolution. This polyploid characteristic leads to the complex interaction between genes, which increases the difficulty of gene function analysis, and also provides rich genetic material for the emergence of new traits.
Chromosome structural variation: There are a lot of chromosome structural variations in wheat genome, such as inversion, translocation, deletion and repetition. These structural variations may affect the arrangement order and expression regulation of genes, and even lead to recombination inhibition between chromosomes, thus affecting genetic diversity and breeding process.
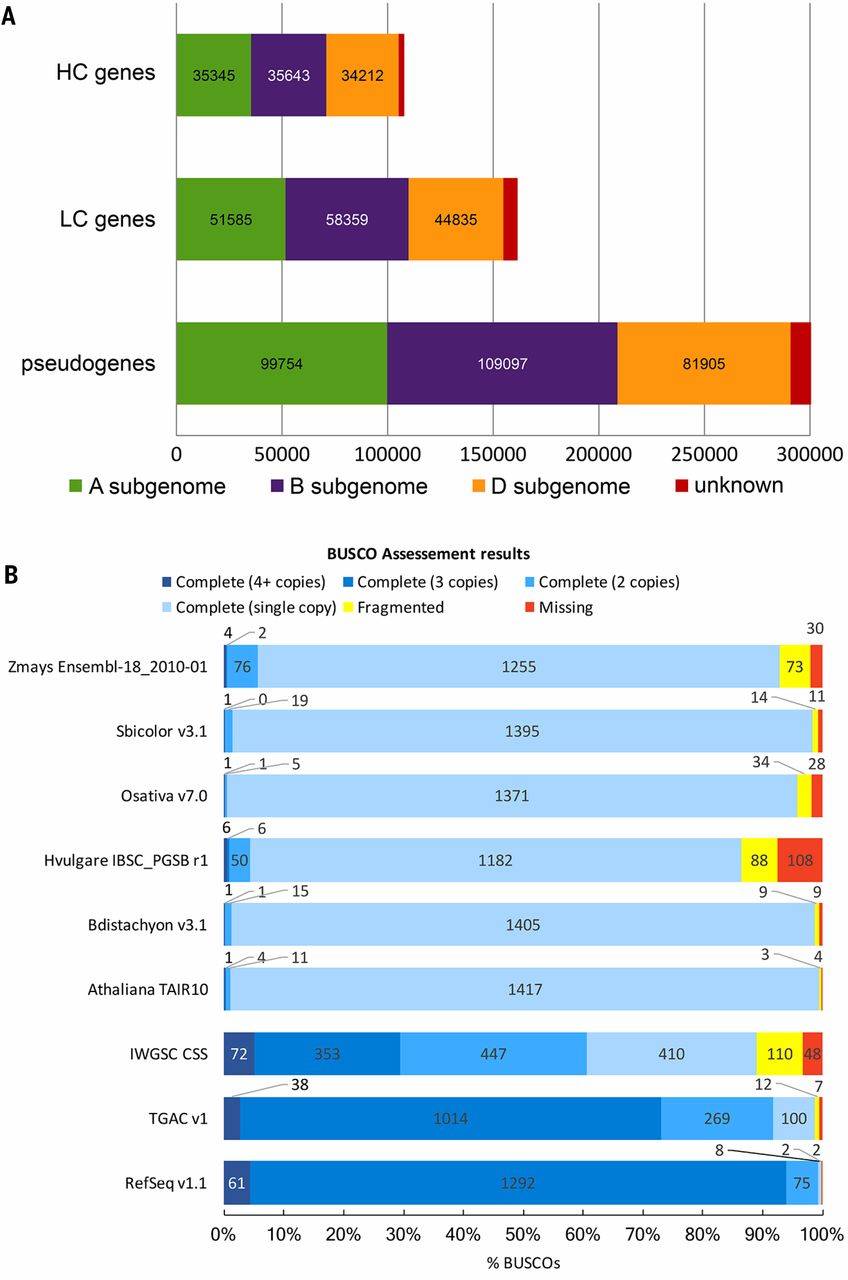
Research Method
Long reading and sequencing technology: The emergence of long reading and long sequencing technology has greatly promoted the study of complex regions of wheat genome. Long reading and sequencing can span repetitive sequence regions and assemble chromosome fragments accurately, which fills the gap of short reading and long sequencing in complex region assembly. Through long reading and sequencing, researchers successfully analyzed the sequence structures of some highly repetitive centromere regions and telomere regions in the wheat genome, laying a foundation for further understanding of chromosome functions.
Optical mapping and Hi-C technology: Optical mapping technology can obtain high-resolution physical mapping by digesting and imaging a single DNA molecule with restriction endonuclease, which can assist genome assembly and structural variation detection. Hi-C technology is based on chromatin conformation capture, which can study the three-dimensional spatial structure of chromosomes at the whole genome level, reveal the long-distance interaction between gene regulatory elements and target genes, and help to analyze the regulatory network in complex regions.
Genomics technology: By using comparative genomics, comparing the wheat genome with other Gramineae genomes, we can identify the conserved regions and specific regions, and infer the gene function and evolutionary relationship. Functional genomics methods, such as gene editing (CRISPR-Cas9) and RNA interference (RNAi), can verify the functions of candidate genes in complex regions and clarify their roles in wheat growth and character formation.
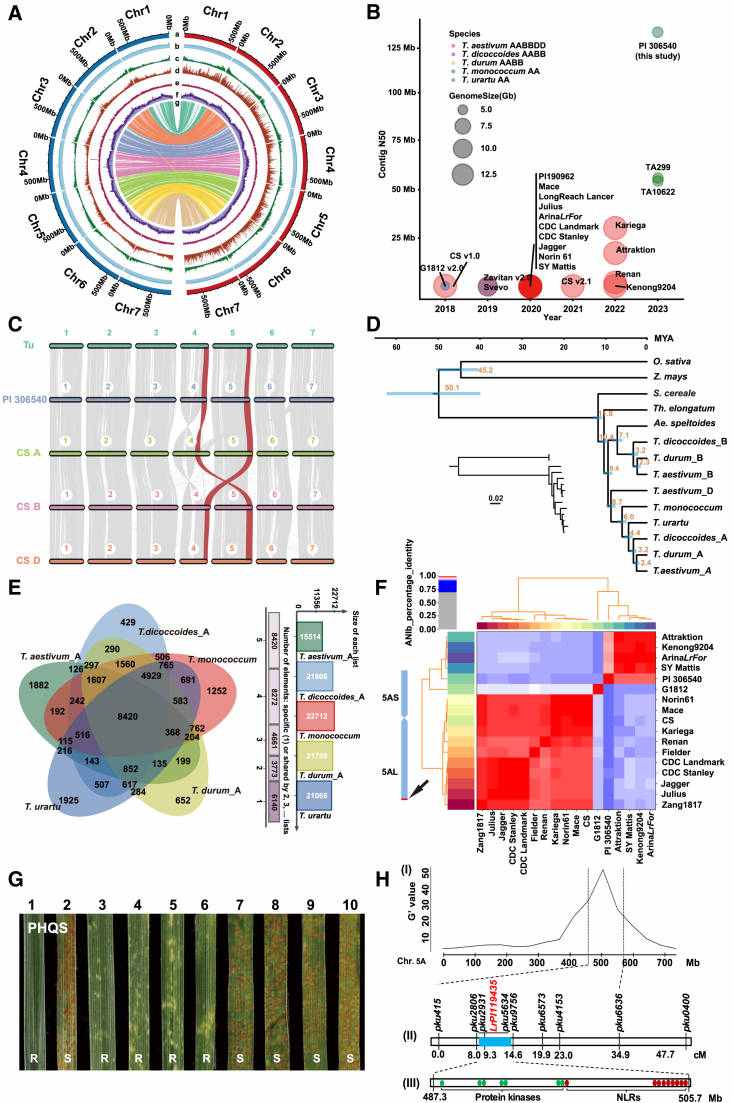
Services you may interested in
Wheat Genome Annotation and Functional Analysis
Wheat genome annotation is a complex and challenging task because of its huge genome, high repetitive sequence content and polyploid characteristics. Despite these difficulties, great progress has been made in recent years, which makes people have a more comprehensive understanding of the wheat genome.
Wheat Genome Annotation
Gene structure annotation: Gene structure annotation aims to determine the position, components and transcription products of genes in the genome. Early annotation of wheat gene structure mainly relied on homologous sequence alignment and prediction algorithm based on machine learning. The application of full-length transcriptome sequencing makes the annotation of wheat gene structure more accurate. These techniques can directly determine the full-length sequence of transcripts, identify alternative splicing events and new subtypes of transcripts, and greatly enrich the structural information of wheat genes.
Functional annotation: Functional annotation is a process that endows genes with biological functions. Common methods include comparing wheat gene sequences with gene databases with known functions (such as nr database of NCBI and Swiss-Prot database) based on sequence similarity comparison (such as BLAST), and inferring gene functions according to similarity. Based on the annotation system of Gene Ontology (GO), the gene function is systematically described from three levels: molecular function, biological process and cell component. And using Kegg (Kyoto encyclopedia of genes and genomes) database to analyze metabolic pathways, so as to clarify the metabolic pathways and regulatory networks in which genes participate. In addition, with the accumulation of protein-protein interaction data, functional annotation can also be assisted by analyzing the interaction network of gene-encoded proteins.
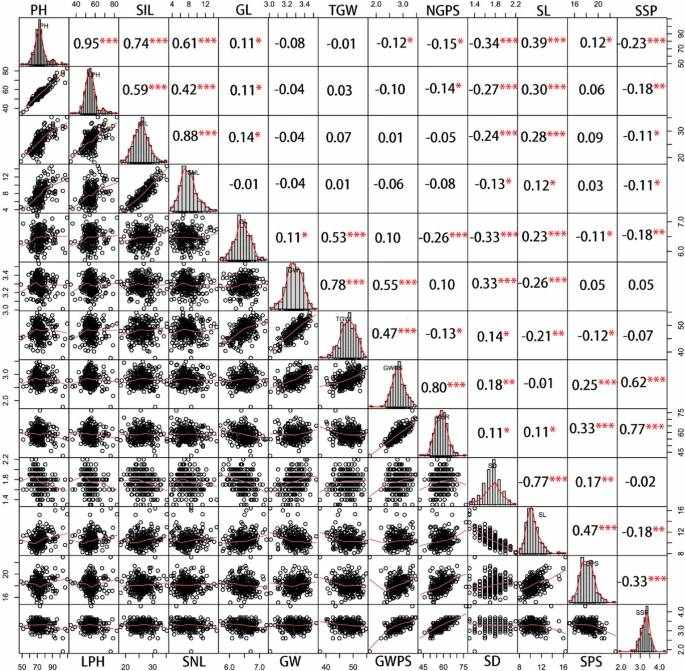 Correlation coecients among yield-related traits calculated from BLUP values (Lv et al., 2024)
Correlation coecients among yield-related traits calculated from BLUP values (Lv et al., 2024)
Functional Analysis of Wheat Genes
Transcriptomics: It studies screen differentially expressed genes by analyzing the expression profiles of wheat genes under different growth and development stages, tissues and organs or environmental stresses, and then mine genes related to specific biological processes or traits.
Metabonomics: It can directly reflect the physiological state of organisms by detecting the changes of wheat metabolites. Combined with transcriptome data, the regulatory relationship between genes and metabolites can be deeply analyzed.
Epigenomics: Epigenetic modifications such as DNA methylation and histone modification play an important role in gene expression regulation. Epigenomics studies revealed the dynamic changes of these modifications in wheat growth and environmental response, as well as their effects on gene function.
Genetic transformation: It is a direct method to verify the gene function by changing the expression level of the target gene in wheat and observing the phenotypic changes through genetic transformation techniques such as gene overexpression, gene knockout or gene silencing. Using CRISPR-Cas9 system to knock out a disease-resistant gene in wheat, the resistance of plants to the corresponding pathogen was significantly reduced, which proved that the gene had an important function in the disease-resistant process of wheat.
Mutant analysis: screening and identifying natural or artificially induced mutants of wheat and analyzing the relationship between mutant phenotype and gene variation are also common means to verify gene function.
Genetic Variation and Trait Association Analysis
Genetic variation and trait association analysis of wheat is the core means to analyze the genetic basis of complex agronomic traits of wheat and promote molecular breeding. Genetic variation is the basis of biological evolution and trait diversity.
Genome-wide Association Study
Based on the linkage disequilibrium principle, GWAS locates the correlation sites of target traits by analyzing the correlation between genetic variation and phenotypic traits in natural populations. The research process includes population structure analysis, phenotypic data collection, genotyping and statistical analysis (commonly used mixed linear model) to correct the influence of population structure and kinship on the results.
In the study of wheat yield traits, GWAS located several key sites for regulating grain number per spike and 1000-grain weight. For quality traits, SNP loci related to gluten content and starch gelatinization characteristics were identified by correlation analysis. In the research of disease resistance, the resistance genes of powdery mildew, rust and other pathogens were successfully located, which provided marker resources for disease resistance breeding.
Linkage Analysis
Based on the genetic map, the linkage relationship between genes and markers was determined by analyzing the co-segregation of markers and traits in the isolated population. Commonly used segregation populations include F2 population, recombinant inbred line (RIL), near isogenic line (NIL) and so on. With the construction of high-density genetic map, the accuracy of quantitative trait loci (QTL) location has been continuously improved.
The genetic map of wheat was constructed by RIL population, and several QTL controlling plant height and heading date were located. Through near-isogenic line analysis, the stripe rust resistance gene Yr27 was finely located and cloned, which provided important materials for functional research and molecular breeding of disease resistance genes.
Other Analytical Methods
Based on the known functional genes or metabolic pathways, the genetic variation in candidate genes was screened and the correlation analysis was carried out with the target traits. This method has strong pertinence and is often used to verify the relationship between genes related to specific biological processes and traits.
Integrating genome, transcriptome, metabolomics and other omics data, the relationship between genetic variation and traits was analyzed from multiple dimensions. Combining with transcriptome data, the association between differentially expressed genes and GWAS loci can be revealed, and the mechanism of gene regulatory network on trait formation can be clarified.
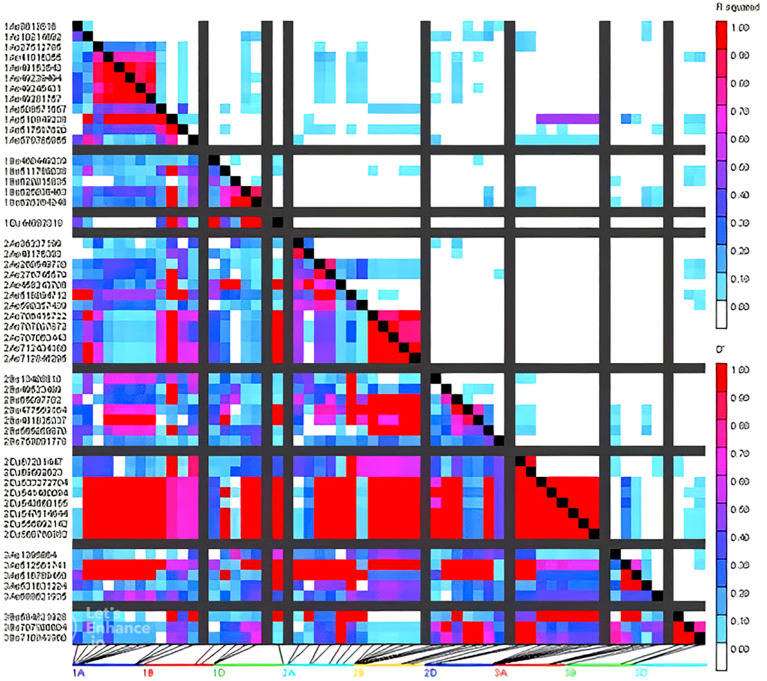 Patterns of LD blocks (right) of GWAS results indicating the position of candidate genes and/or QTL regions associated with grain quality traits and agronomic traits (Vishwakarma et al., 2024)
Patterns of LD blocks (right) of GWAS results indicating the position of candidate genes and/or QTL regions associated with grain quality traits and agronomic traits (Vishwakarma et al., 2024)
Services you may interested in
Wheat Genome Analysis for Evolutionary Events
The rapid development of genomics provides a powerful tool for studying the evolution of wheat. By analyzing the genomes of wheat and its ancestors, key events in the evolution process can be revealed, which is of great significance for understanding the biological characteristics of wheat and improving wheat varieties.
Adaptive Evolutionary Characteristics
By comparing and analyzing the genomes of wheat and its wild ancestors, researcher can identify the genomic regions that were positively selected in the evolution process. The genes contained in these regions are often closely related to the adaptive evolution of wheat. In some wheat varieties grown in arid areas, the genomic regions related to root development and water absorption show obvious adaptive evolution characteristics, and the variation of related genes enables wheat to obtain water better and improve its viability in water-deficient environment.
In addition to the change of gene sequence, transcription regulatory network also plays a key role in the adaptive evolution of wheat. With the change of environment, wheat reshapes the transcription regulatory network by adjusting the expression and regulatory relationship of transcription factors, thus regulating the expression of downstream genes and realizing adaptation to the environment. This adaptive change of transcription regulatory network enables wheat to quickly adjust its physiological state and maintain its growth and development under different environmental conditions.
Genetic Selection in the Process of Improvement
The development of modern breeding technology has further accelerated the genetic improvement of wheat. By means of hybridization, mutation and molecular marker-assisted selection, breeders make directional selection for high yield, high quality and disease resistance. In this process, many important genes were aggregated into excellent varieties.
By introducing rust-resistant genes, the resistance of wheat to rust was significantly improved. Improve the genes related to starch synthesis and improve the processing quality of wheat. By comparing and analyzing the genomes of modern wheat varieties and ancient wheat varieties, we can clearly see the imprint of these genetic selection and reveal the law of gene changes in the process of modern breeding.
Research Methods and Techniques
Bioinformatics plays a central role in the analysis of wheat genome evolution. Through sequence comparison, gene family identification, selection pressure analysis and other methods, we can identify the evolutionary relationship of genes, analyze the expansion and contraction of gene families, and detect selected genomic regions. At the same time, using transcriptomics, protein omics and other omics data, combined with bioinformatics analysis, we can deeply study the expression regulation and function of genes and reveal the molecular mechanism of wheat evolution.
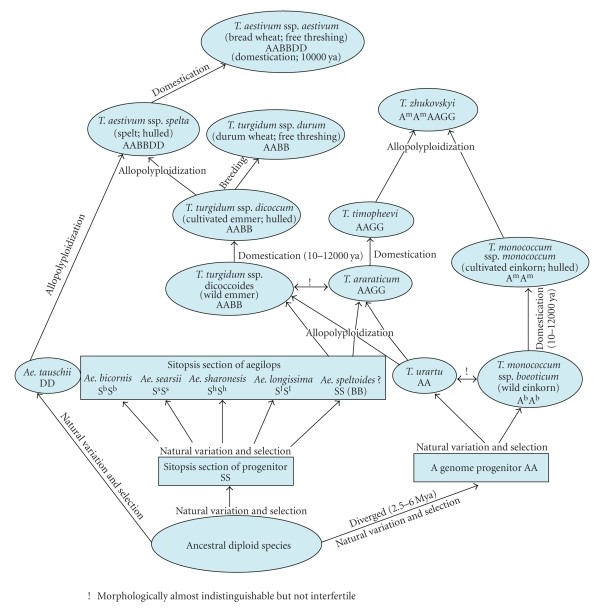 Schematic representation of the evolutionary history of wheat species (Gupta et al., 2008)
Schematic representation of the evolutionary history of wheat species (Gupta et al., 2008)
Services you may interested in
Conclusion
Wheat genome analysis is the important in crop improvement. By integrating multi-omics data and advanced bioinformatics technology, researchers can systematically analyze the regulatory network and evolutionary trajectory of wheat complex genome, and provide accurate molecular targets for directional breeding. From the mapping of variation of global germplasm resources to the functional verification of important agronomic traits genes, wheat genome research is promoting the paradigm shift of breeding from traditional experience-driven to accurate data design.
References
- Wang X, Li H., et al. "A near-complete genome sequence of einkorn wheat provides insight into the evolution of wheat A subgenomes." Plant Commun. 2024 5(5):100768 https://doi.org/10.1016/j.xplc.2023.100768
- International Wheat Genome Sequencing Consortium (IWGSC). "Shifting the limits in wheat research and breeding using a fully annotated reference genome." Science. 2018 361(6403):eaar7191 https://doi.org/10.1126/science.aar7191
- Lv, Y, Dong, L., et al. "Single- and multi-locus genome-wide association study reveals genomic regions of thirteen yield-related traits in common wheat." BMC Plant Biol. 2024 24 1228 https://doi.org/10.1186/s12870-024-05956-y
- Thind AK, Wicker T., et al. "Chromosome-scale comparative sequence analysis unravels molecular mechanisms of genome dynamics between two wheat cultivars." Genome Biol. 2018 19(1):104 https://doi.org/10.1186/s13059-018-1477-2
- Vishwakarma MK., et al. "Genetic dissection of value-added quality traits and agronomic parameters through genome-wide association mapping in bread wheat (T. aestivum L.)." Front Plant Sci. 2024 15:1419227 https://doi.org/10.3389/fpls.2024.1419227
- Gupta PK, Mir RR., et al. "Wheat genomics: present status and future prospects." Int J Plant Genomics. 2008:896451 https://doi.org/10.1155/2008/896451


 Send a Message
Send a MessageFor any general inquiries, please fill out the form below.


