
Decoding the Wheat Genome: New Discoveries and Applications
Wheat is an important food crop in the world, and its genome research has been paid close attention to. Wheat genome is huge, complex and has many repetitive sequences, which makes the analysis of wheat genome very challenging. However, in recent years, with the continuous development of sequencing technology and the unremitting efforts of researchers, a series of remarkable recent progress has been made in wheat genome research. These advances provide an important basis and opportunity for understanding the genetic characteristics and evolutionary mechanism of wheat and carrying out molecular design and breeding.
This paper introduces many achievements in wheat genome research, including revealing multi-locus co-evolution, assisting breeding and functional research, mining genes regulating grain size, drawing T2T genome map of hexaploid wheat, and promoting wheat subgenomic differentiation.
Reveal Multi-locus Co-evolution of Wheat Genome
The researchers sequenced the exons of 287 wheat materials. Population genetic analysis shows that 6.7% of wheat genome is located in the selective scanning area of local varieties and cultivated varieties, which contains known genes to improve yield. These regions are asymmetrically distributed on A and B subgenomes, which is beneficial to the selection of regulatory genes. Genome-wide association studies (GWAS) identified genomic loci related to yield potential traits, and located and characterized two pleiotropic genes: TaARF12 encodes auxin-related factors, and TaDEP1 encodes G protein γ subunit, which together regulate plant height and grain weight.
Asymmetric Selection Between Subgenomes
By comparing the plant height and the phenotypic average value of seeds, it was found that the plant height of cultivated varieties was significantly lower than that of local varieties, but the grain length (GL), grain thickness (GT), grain width (GW) and 1000-grain weight (TGW) were significantly higher than those of local varieties. Based on SNP data, selection signal detection and analysis (Fst, Pi, XP-CLR) were carried out, and 975.4Mb selection intervals were obtained. In these intervals, 37 previously reported genes in rice were found, such as plant height-related gene d2 and grain weight-related gene TaSus2-2A. It is also found that most of these intervals come from A and B subgenomes. By using triplet genes to study this asymmetric selection, the results showed that 2204, 1265 and 300 genes were specifically selected in A, B and D subgenomes, respectively.
Through further gene function enrichment analysis, it was found that among the 34 auxin-related genes, 24 auxin-related response genes came from three sub-genomes of ABD, and the remaining 10 genes involved in upstream function regulation came from A and B sub-genomes. In D sub-genome, signal transduction genes were rarely selected. This indicates that breeding may give priority to upstream genes.
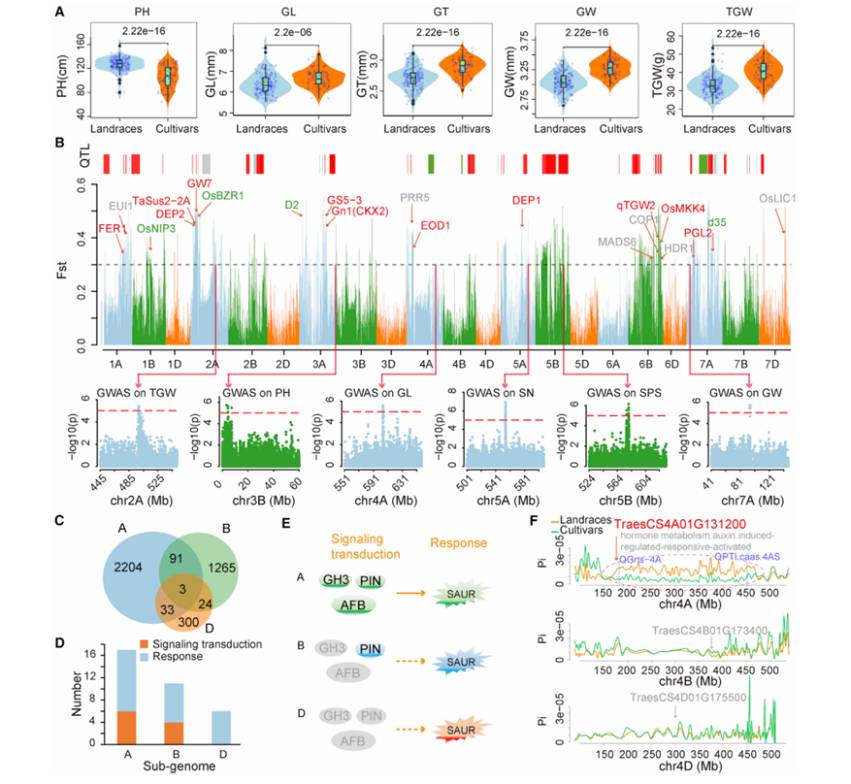 Selection signatures for plant height and yield traits between cultivars and landraces (Li et al., 2022)
Selection signatures for plant height and yield traits between cultivars and landraces (Li et al., 2022)
Identification of Efficient Genes by GWAS Analysis
Genome-wide association study correlation with plant height phenotype identified 10 genomic loci, one of which was located at the end of chromosome 2A with only one peak. The study found that plants containing TT SNH (single nucleotide haplotype) had shorter plant height, and at the same time, the grain length, grain width, grain thickness, 1000-grain weight and flowering time increased, which was identified as a typical pleiotropic locus.
Further genome-wide scanning within 3Mb of SNPs peak found 11 genes expressed in wheat stems. Among these 11 genes, TraesCS2A01G547800 was abundantly expressed in the early stage of stem development, and it was the only gene that was significantly down-regulated during stem development, and it was the homologous gene of known plant height gene DLT (plant height correlation) in rice. Because it encodes auxin response factor, it is named TaARF12. Under the background of Rht-1, the expression of TaARF12 was down-regulated by RNAi technology, and the plant height of transgenic plants was significantly reduced. At the same time, the ears of transgenic plants were obviously longer, and the seed setting rate per ear was significantly increased.
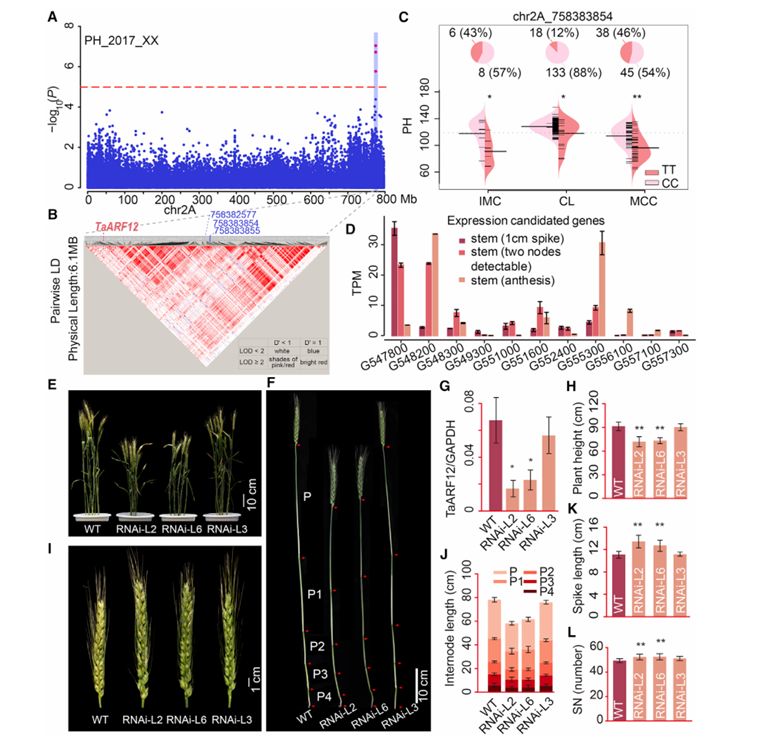 Identification of a novel major locus for plant height on chromosome 2A (Li et al., 2022)
Identification of a novel major locus for plant height on chromosome 2A (Li et al., 2022)
Services you may interested in
Help Wheat Breeding and Functional Research
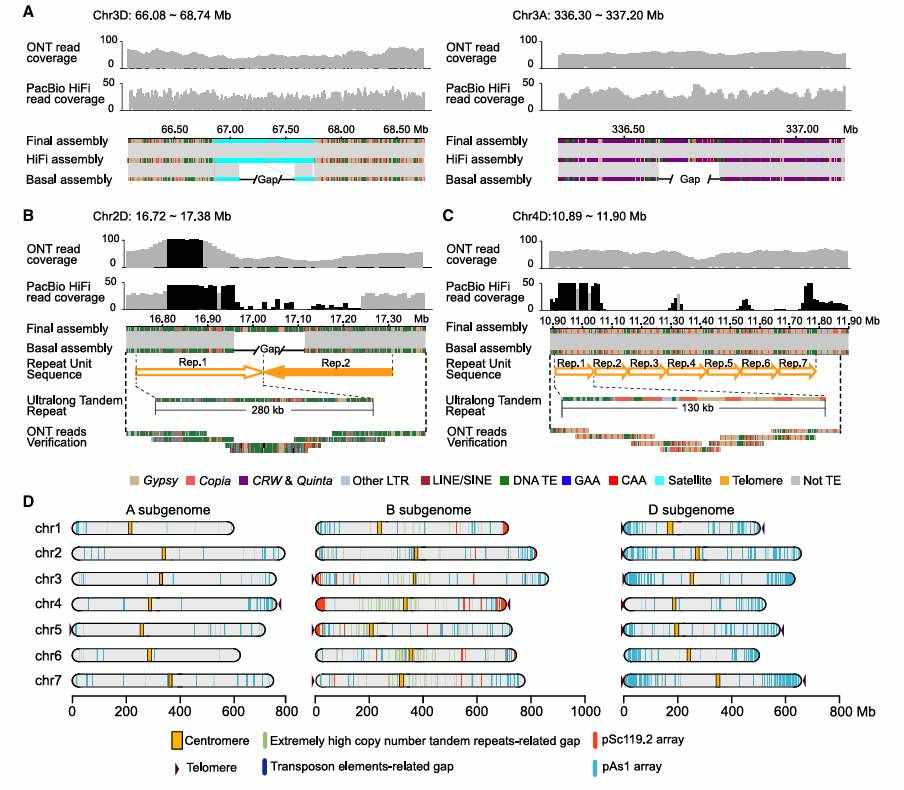
Using ONT long reading length sequencing (coverage 283.5×), PacBio HiFi high-precision sequencing (29.01×) and Hi-C data, the researchers realized the near-complete assembly (CS-CAU) of wheat China Spring genome, with a size of 14.46 Gb, a base accuracy of more than 99.9963%, and only 290 assembly gaps (mainly ultra-long tandem repeats). Among them, 1D, 3D, 4D and 5D chromosomes assembled without gaps for the first time, and 1D and 5D chromosomes reached telomere-to-telomere (T2T) level. This breakthrough not only solves the assembly problem of high repetitive sequence and complex polyploid of wheat genome, but also provides a model for analyzing other complex crop genomes.
 Near-complete assembly of the Chinese Spring genome (Wang et al., 2025)
Near-complete assembly of the Chinese Spring genome (Wang et al., 2025)
Based on the near-complete genome assembly, the research team annotated a total of 151,405 high-confidence genes, of which 59,180 were newly annotated genes, including 7,602 genes assembled for the first time, which is of great significance to the study of wheat gene function. By integrating RNA-seq data set and cross-species protein homology evidence, the genome distribution and expression characteristics of six kinds of seed storage proteins (SSPs) were completely analyzed for the first time. It was found that the expression of ω-gliadin was entirely contributed by B subgenome, while the expression of other five types of SSPs (α/γ-gliadin, ALP, HMW/LMW glutenin) was mainly contributed by D subgenome, which provided an important basis for further analyzing the genetic basis and molecular improvement of wheat gluten quality.
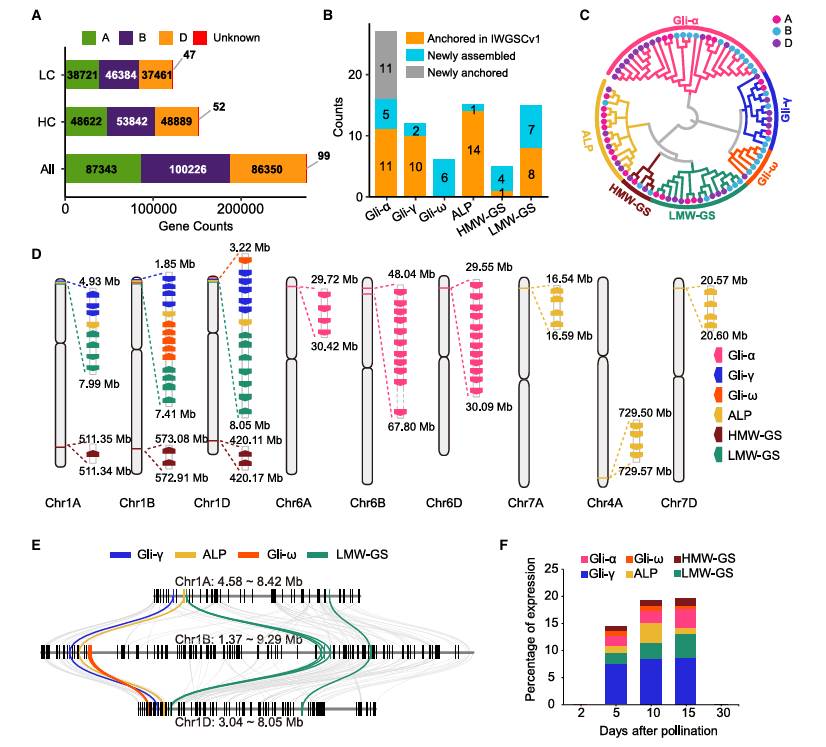 Annotation of the near-complete CS genome (Wang et al., 2025)
Annotation of the near-complete CS genome (Wang et al., 2025)
The centromere sequences of the other 20 chromosomes were all assembled except the centromere of chr1B, which had gaps related to the long GAA repeats. The analysis of the sequence composition of centromere region shows that the centromere region is mainly composed of retrotransposons, in which the centromere of A/B subgenome is rich in centromere-related retrotransposons CRW and Quinta (accounting for about 70%), while only 30% of the centromere of D subgenome is composed of CRW and Quinta.
Similarly, the distribution of tandem repeats among the three subgenomes is highly uneven, in which 71.89% of simple tandem repeats (SSR) are enriched in the B subgenome, while nearly half of satellite sequences are concentrated in the D subgenome. In addition, the research team also analyzed the insertion time of CRW and Quinta transposons in centromere region, and made clear the main expansion period among the three subgenomes.
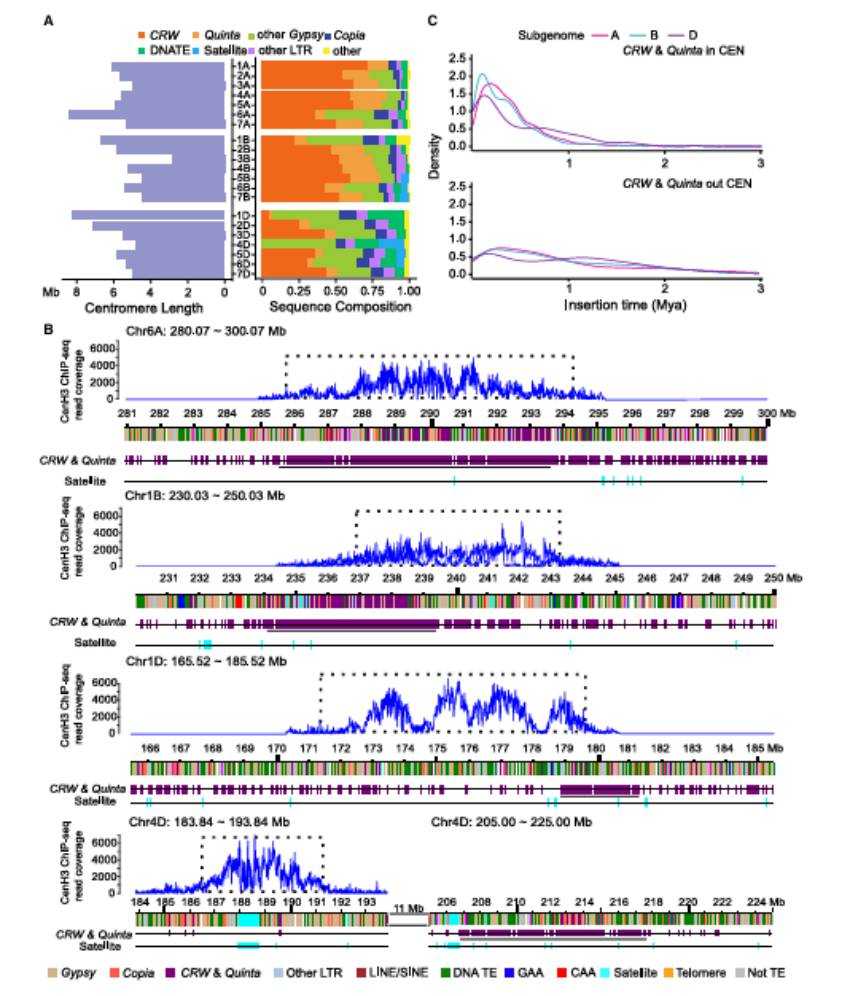 The sequence constitution and insertion time of CRW and Quinta in centromeric regions (Wang et al., 2025)
The sequence constitution and insertion time of CRW and Quinta in centromeric regions (Wang et al., 2025)
Gene Mining of Wheat Grain Size Regulation
In order to mine the genes regulating the grain size of wheat, the researchers investigated the grain size and 1000-grain weight of 349 wheat materials for many years. Through GWAS analysis, a Block with significant multi-environment was identified, which contained 11 candidate genes. Furthermore, TaSRK was locked as the key candidate gene by combining EMS mutant library, transcription expression and gene sequencing information of wheat Pan800K. There were four haplotypes of this gene, among which TaSRK-Hap1 had significantly higher grain length and 1000-grain weight. The field data for three consecutive years showed that the grain length and 1000-grain weight of transgenic wheat overexpressed by TaSRK became shorter, while the mutant showed that the grain length became longer and the 1000-grain weight increased significantly.
Using TaSRK protein as bait, the wheat cDNA library was screened by yeast two-hybrid method, and a histone deacetylase TaHDA9 was determined. The experiment confirmed that there was protein interaction between TaSRK and TaHDA9. In vitro and semi-in vitro deacetylation experiments confirmed that TaHDA9 directly deacetylated two lysine sites K541 and K733 of TaSRK, thus reducing its protein accumulation. The investigation results of agronomic characters for three consecutive years from 2022 to 2024 showed that the grain length and 1000-grain weight of TaHDA9 overexpressed transgenic wheat increased significantly, while the mutant grain length and 1000-grain weight decreased significantly.
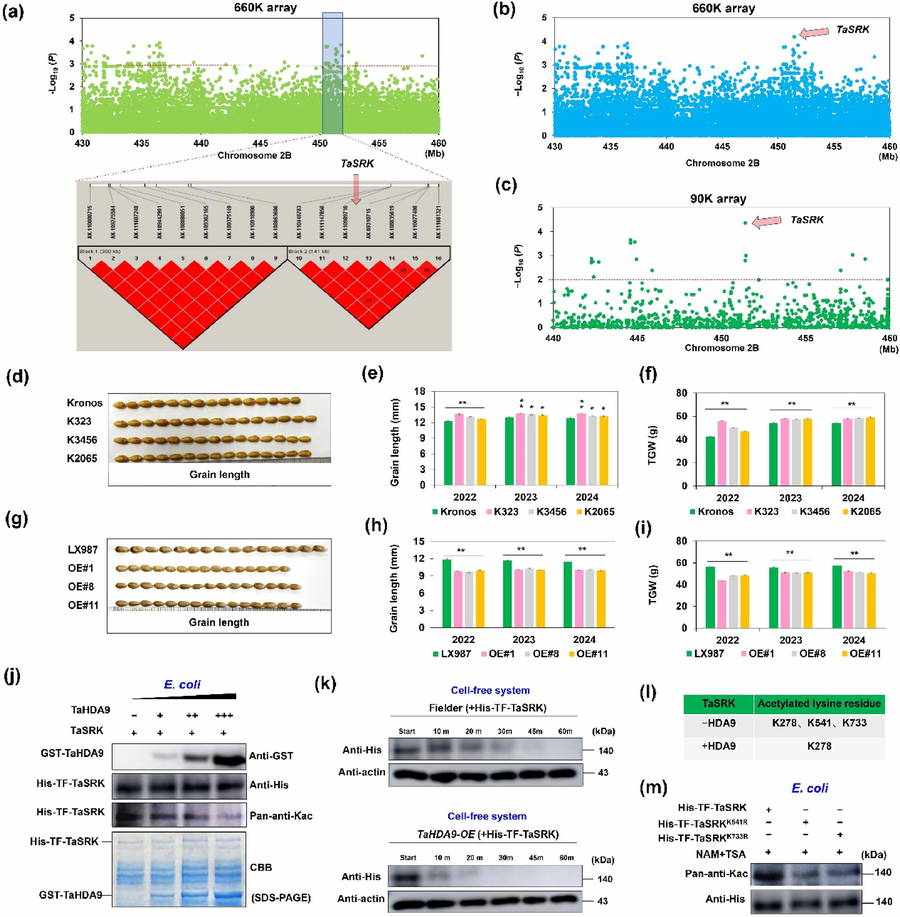 Cloning and functional identification of TaSRK (Zhang et al., 2025)
Cloning and functional identification of TaSRK (Zhang et al., 2025)
In order to clarify the mechanism of TaSRK regulating wheat grain length, we also focused on another sieve library protein TaPsbO. The experiment confirmed that TaSRK interacted with TaPsbO in chloroplast, which indicated that TaSRK might play its role by interacting with TaPsbO in chloroplast. The results of phosphorylation in vitro and in vivo showed that TaSRK could directly phosphorylate seven sites (including key site S222) of TaPsbO(TaPsbOS222) and reduce its protein abundance.
Haplotype analysis shows that TaPsbO has a rare haplotype TaPsbOG222 in nature. The results of protein interaction showed that TaSRK and TaPsbOG222 had stronger interaction. In vitro and in vivo phosphorylation experiments confirmed that TaSRK had differential phosphorylation regulation on TaPsbOS222 and TaPsbOG222, and the haplotype protein abundance of TaPsbOG222 was higher.
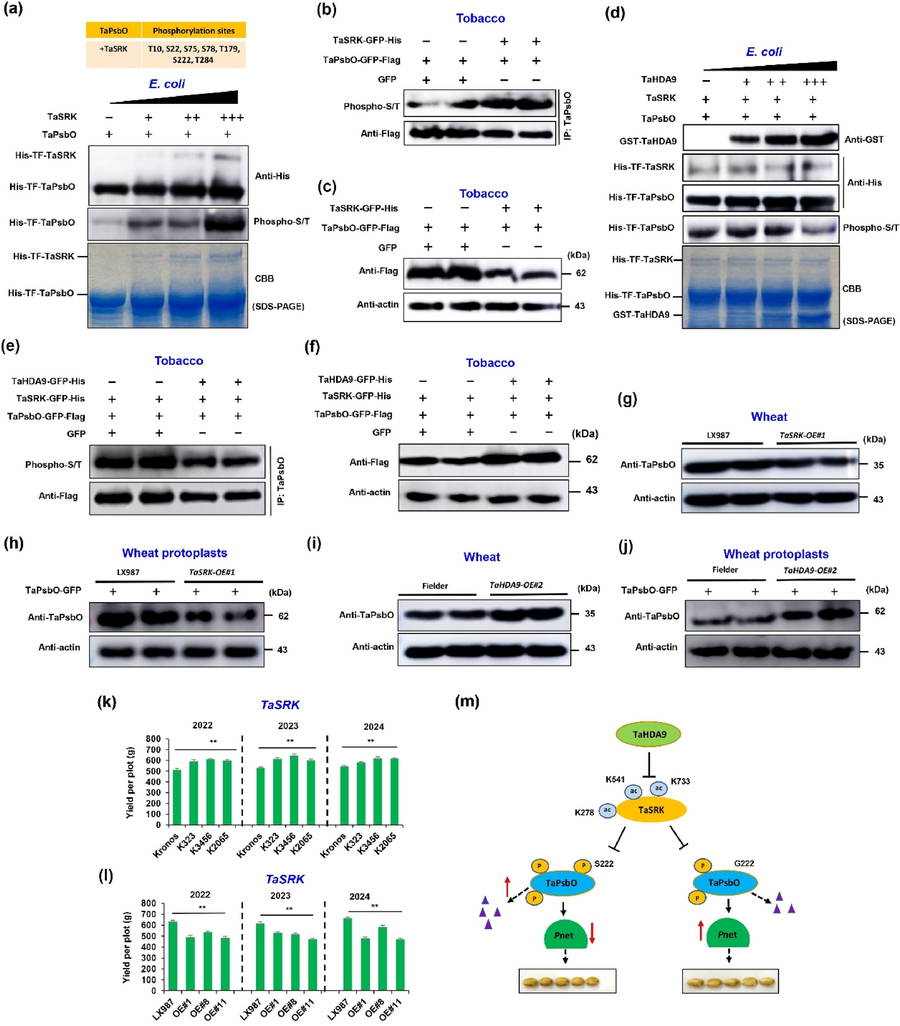 TaHDA9-TaSRK-TaPsbO module regulates wheat grain length by mediating photosynthesis (Zhang et al., 2025)
TaHDA9-TaSRK-TaPsbO module regulates wheat grain length by mediating photosynthesis (Zhang et al., 2025)
Draw Complete Genome Map of T2T of Hexaploid Wheat
Recently, the wheat genome map has made a breakthrough: the telomere-to-telomere (T2T) complete genome map of hexaploid wheat was successfully drawn for the first time in the world, and the precise assembly of the wheat genome from the "head" to the "tail" was realized.
A Variety of High-precision Sequencing Combination Strategies
The research team successfully constructed the T2T genome of hexaploid wheat, named CS-IAAS version 1.0, by using cutting-edge technologies such as high-precision sequencing of PacBio HiFi and ONT ultra-long reading length sequencing, combined with various algorithms. The total length of the genome reached 14.51 Gb (about 14.5 billion bases), and the seamless splicing of 42 wheat chromosomes from telomere to telomere was realized for the first time. Compared with the past, this map has achieved a qualitative leap in completeness, continuity and accuracy, laying a solid foundation for functional genomics research. This achievement shows China's leading position in the field of agricultural genomics research and provides strong scientific and technological support for food security strategy.
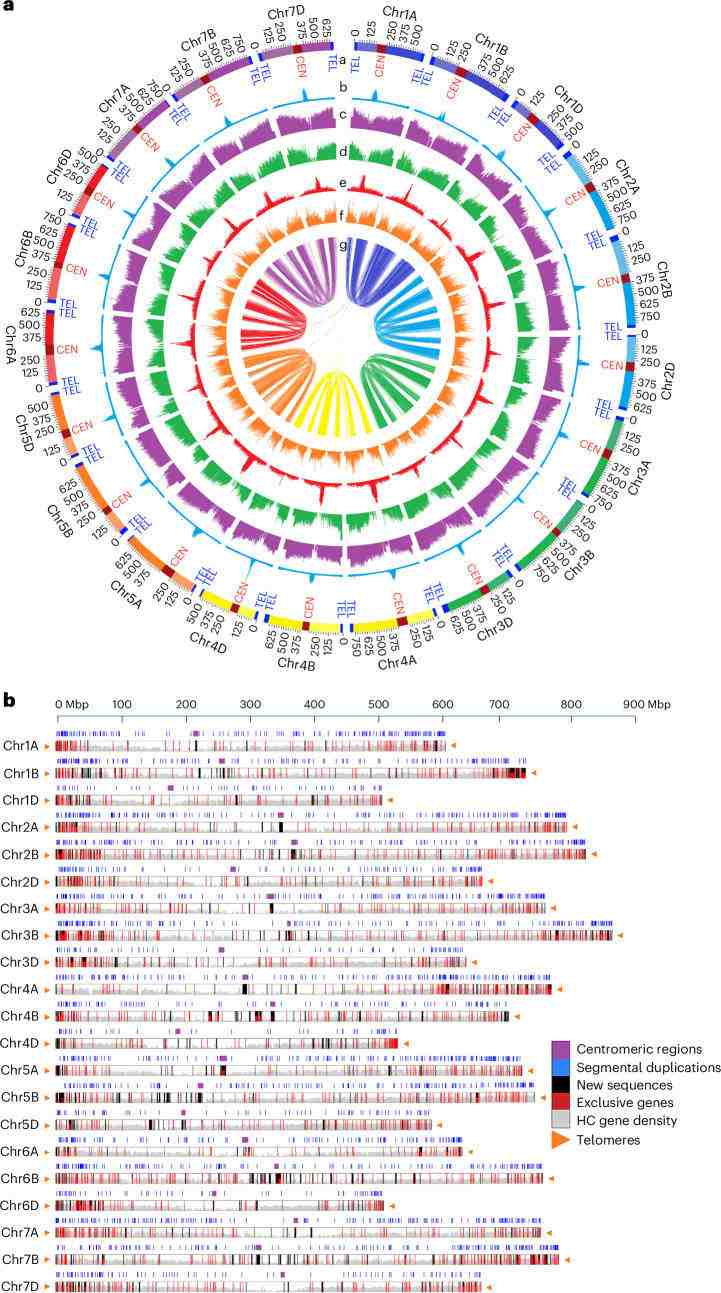 Summary of the complete CS-IAAS genome assembly (Liu et al., 2025)
Summary of the complete CS-IAAS genome assembly (Liu et al., 2025)
Uncover the Secrets of Complex Regions of Chromosomes
With the help of the complete genome map, the research team clearly analyzed the complex regions such as centromeres, telomeres and ribosomal DNA repeats (rDNA arrays) in the wheat genome for the first time. Modern common wheat is a hexaploid crop formed by crossing three ancestor species. Using T2T genome map, the research team revealed 23 major chromosome segment inversions in the process of wheat evolution from tetraploid to hexaploid, with a total length of about 518 million bases. These inversions are all preserved in modern wheat, and the breakpoints are rich in special short repetitive sequences, which presumably play a role in chromosome breakage and reconnection.
The release of telomere-to-telomere complete genome map of hexaploid wheat marks a new stage of wheat genome research. This achievement not only deepens the understanding of wheat genome structure and evolutionary mechanism, but also provides an example for analyzing the genomes of other complex polyploid crops. Relying on this high-quality reference genome, scientists will more accurately mine key genes related to yield, quality and disease resistance, and bring revolutionary breakthroughs to wheat variety improvement.
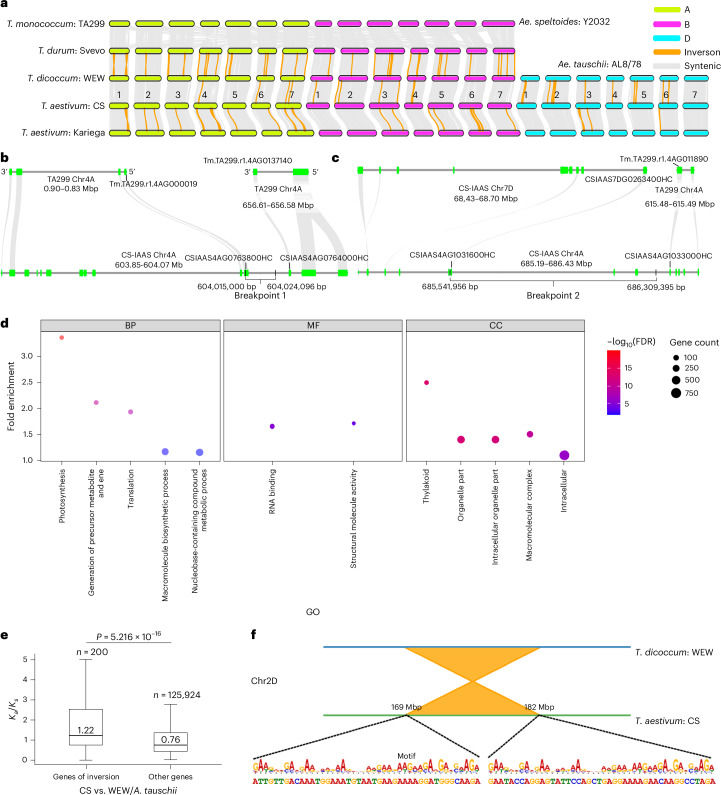 Chromosomal rearrangements contribute to the evolution of hexaploidization in wheat (Liu et al., 2025)
Chromosomal rearrangements contribute to the evolution of hexaploidization in wheat (Liu et al., 2025)
Driving Wheat Subgenomic Differentiation
Nearly 85% of the genome of hexaploid wheat is composed of transposable element TE. Although the three subgenomes of wheat, A, B and D, are similar in the overall level of TE content, about 42% of TE families are biased among subgenomes. In addition, about 95% of the genes are co-located with TEs, and these changeable TEs can lead to copy number variation (CNV), structural variation (SV), methylation variation (MV) and expression variation (EV) of orthologous gene between subgenomes, which greatly promotes the differentiation between subgenomes and homologous genes. It was also found that about 50% of the genes in the wheat genome showed copy number variation, and about 80% of the remaining single copy subgenomic orthologous genes (Triads) showed expression or subfunctional differentiation, including many known genes such as domesticated genes Btr1 and Q and improved genes Ppd1 and Rht1.
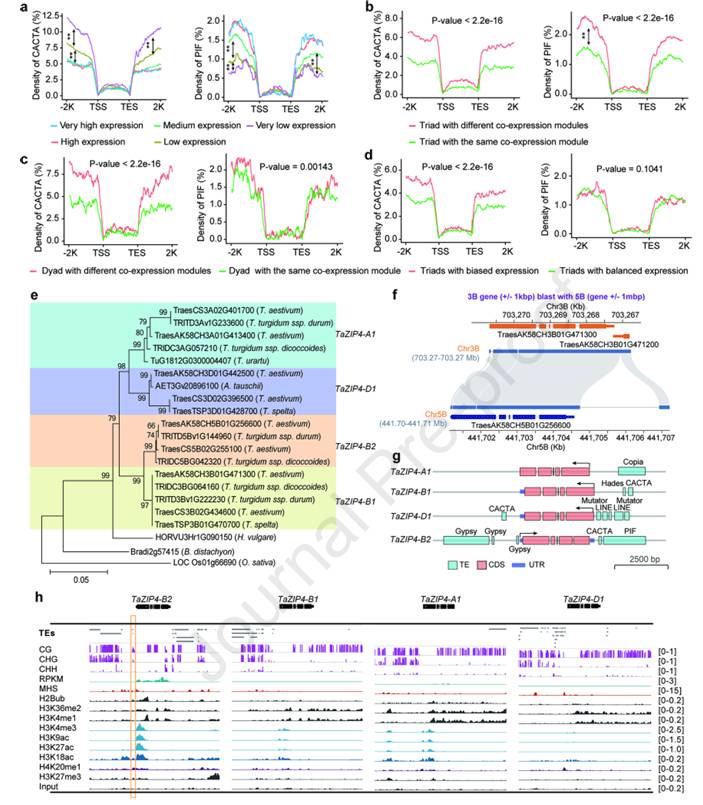 TE mediates subgenomic homologous gene differentiation in wheat (Jia et al., 2025)
TE mediates subgenomic homologous gene differentiation in wheat (Jia et al., 2025)
Compared with diploids, subgenomic homologous genes of hexaploid species show potential neo-functionalization or sub-functionalization, which makes polyploid a natural "Heterozygote". These homologous genes are similar to the alleles in diploid heterozygotes, so they are also called "partial homologous alleles". Compared with allelic variation in diploid, excellent variation polymerization in polyploid involves not only different gene loci, but also some homologous genes in subgenome. This feature enables polyploids to express excellent partial homozygous haplotypes (EHH) in key spatio-temporal stages, thus playing an important role in long-term natural and artificial selection. Generally speaking, the differentiation of homologous genes between subgenomes has brought significant advantages to polyploidy by increasing the diversity and expression plasticity of functional genes.
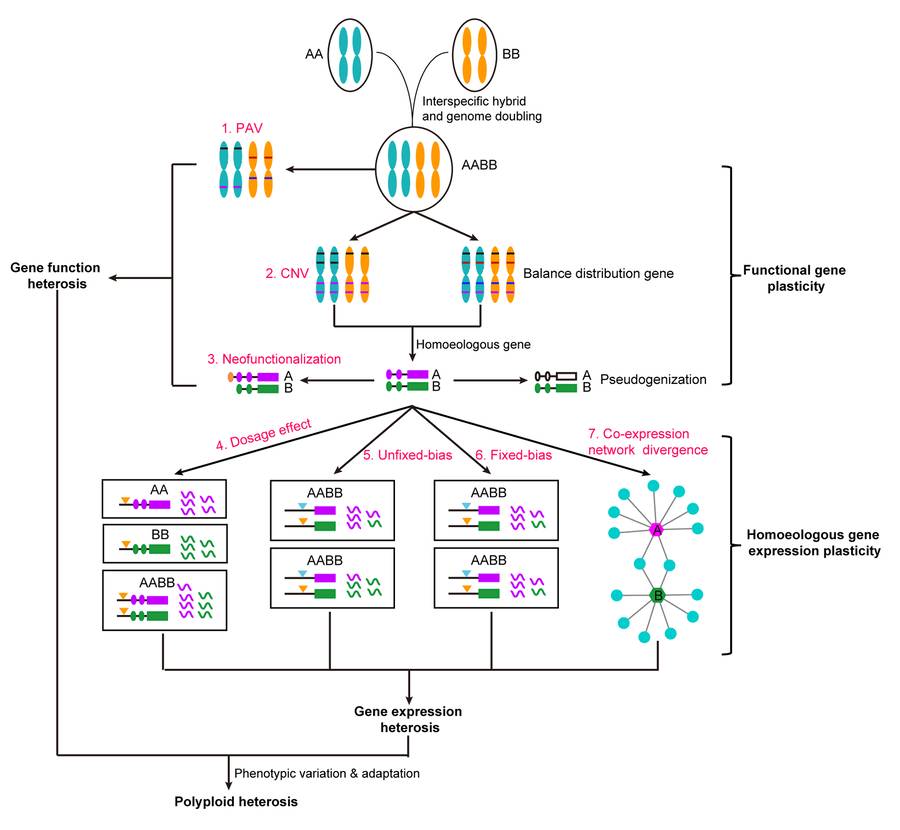 Possible regulation mechanism of polyploid heterosis (Jia et al., 2025)
Possible regulation mechanism of polyploid heterosis (Jia et al., 2025)
Conclusion
To sum up, wheat genome research is in the golden age of rapid development. With the breakthrough of the long read sequencing technology and the innovation of bioinformatics analysis methods, many cutting-edge achievements are constantly emerging. The birth of the world's first telomere-to-telomere complete genome map of hexaploid wheat, based on the deep integration of high-precision nanopore sequencing and chromosome conformation capture technology, seems to light a bright light for the field of wheat research and completely illuminate the research blind spot that once existed because of the complexity and huge genome. This breakthrough is a milestone.
References
- Li A, Hao C., et al. "Wheat breeding history reveals synergistic selection of pleiotropic genomic sites for plant architecture and grain yield." Mol Plant. 2022 15(3):504-519 https://doi.org/10.1016/j.molp.2022.01.004
- Wang Z, Miao L., et al. "Near-complete assembly and comprehensive annotation of the wheat Chinese Spring genome." Mol Plant. 2025 S1674-2052(25)00068-1 https://doi.org/10.1016/j.molp.2025.02.002
- N. Zhang, S. Li., et al. "A lectin receptor-like kinase TaSRK that is deacetylated by TaHDA9 regulates wheat grain length by mediating the Photosystem II protein TaPsbO." Science Bulletin (2025) https://doi.org/10.1016/j.scib.2025.04.028
- Liu S, Li K., et al. "A telomere-to-telomere genome assembly coupled with multi-omic data provides insights into the evolution of hexaploid bread wheat." Nat Genet. 2025 57(4):1008-1020 https://doi.org/10.1038/s41588-025-02137-x
- Jia, J., Deng., et al. "Transposon elements drive subgenomic divergence of homoeologous genes to shape wheat domestication and improvement." PLANT COMMUNICATIONS (2025) https://doi.org/10.1016/j.xplc.2025.101341



 Send a Message
Send a MessageFor any general inquiries, please fill out the form below.


