
A Guide to Different RAD-seq Sequencing Technologies: Comparison and Applications
Since its emergence in 2008, RAD-seq technology has given rise to numerous variants. These differ in enzyme digestion strategies, fragment selection methods, library construction processes, and application scenarios. This article aims to clarify the principles and applications of various RAD-seq technologies and offers guidance on selecting the appropriate RAD-seq method for different situations.
Overview of RAD-seq Technologies
RAD-seq (Restriction Site-Associated DNA Sequencing) is a high-throughput genome sequencing technique that utilizes restriction enzyme cleavage sites to generate randomly distributed markers across the genome. As an efficient method for obtaining high-density SNP data across numerous individuals, it has been widely applied in evolutionary biology and ecology, including genetic diversity analysis, genetic linkage mapping, population genetics, and speciation studies. This approach enables systematic genome-wide analysis by targeting loci adjacent to restriction sites, providing cost-effective solutions for non-model organisms without requiring prior genomic resources. First introduced in a 2008 PLOS ONE publication titled "Rapid SNP Discovery and Genetic Mapping Using Sequenced RAD Markers", the original paper has accumulated over 3,000 citations (Google Scholar) and inspired subsequent development of multiple derivative techniques based on restriction endonucleases.
RAD-seq is a reduced-representation genome sequencing technique based on restriction enzymes. Its core principle involves three key steps: enzymatic digestion, fragment selection, and sequencing, which efficiently obtain genome-wide genetic markers (e.g., SNPs) while being particularly suitable for species lacking reference genomes. Depending on the types and quantities of restriction enzymes employed, RAD-seq can be categorized into various technical approaches including Original RAD-seq, ddRAD-seq (Double Digest Restriction-site Associated DNA Sequencing), GBS (Genotyping-by-Sequencing), 2b-RAD-seq (Type IIB Restriction-site Associated DNA Sequencing), and ezRAD (Enzyme-free Restriction-site Associated DNA Sequencing). Due to its pooling-based library construction strategy, RAD-seq technology enables simultaneous preparation of up to 96 sequencing libraries in a single experiment, demonstrating significantly greater operational convenience compared to Paired-end and Mate-pair library preparation methods.
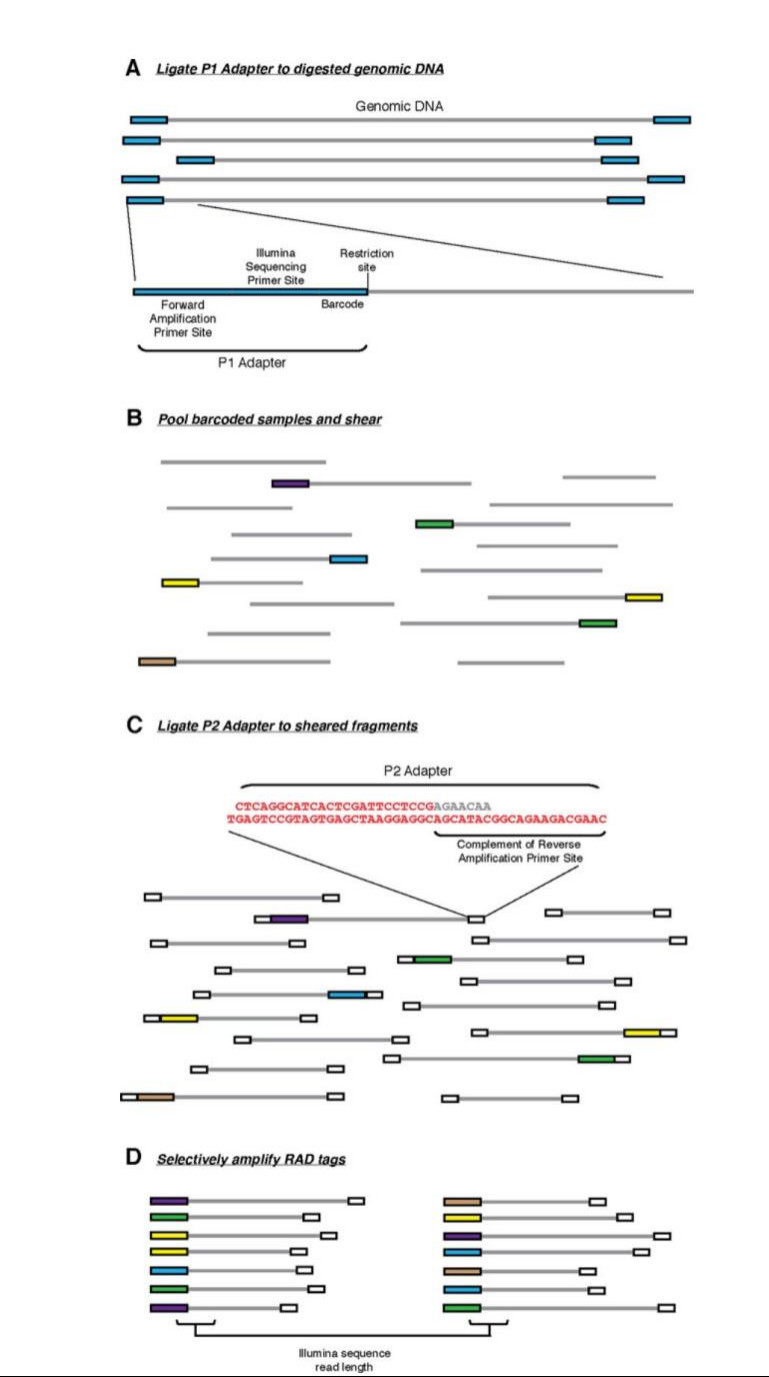 RAD marker generation (Baird, N. A et al.,2008)
RAD marker generation (Baird, N. A et al.,2008)
Comparison of Different RAD-seq Technologies
The primary variants of RAD-SEQ include Original RAD-seq, GBS, ddRAD-seq, 2bRAD, and ezRAD. Each methodology differs in digestion approach, fragment selection criteria, and application scenarios. For instance, Original RAD-seq employs single-enzyme digestion followed by random fragmentation, whereas ddRAD-seq utilizes dual-enzyme digestion with size selection, resulting in superior library uniformity. GBS features a simplified workflow but may exhibit lower marker density. The following section provides a brief overview of each technique.
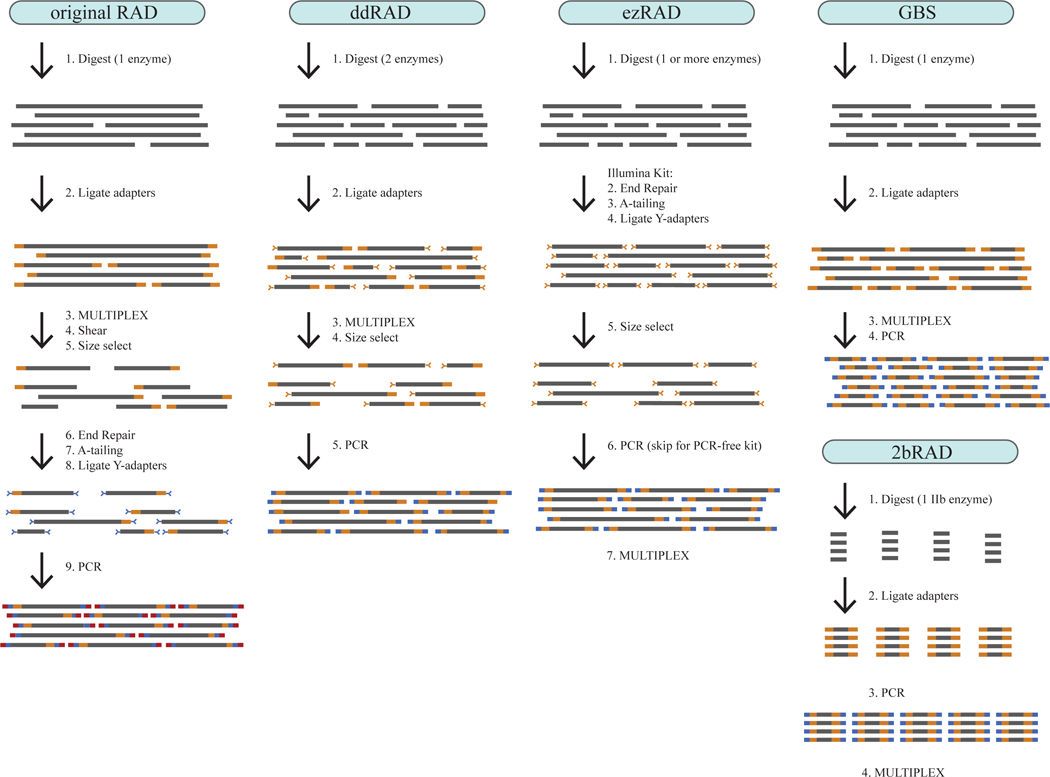 Step-by-step illustration of five RADseq library prep protocols (Kimberly R et al.,2016)
Step-by-step illustration of five RADseq library prep protocols (Kimberly R et al.,2016)
Services you may interested in
Technical analysis of precise fragment preparation in ddRAD-seq
Double digestion: ddRAD-seq simplifies genomic complexity by digesting genomic DNA with two restriction enzymes. These enzymes recognize distinct DNA sequences and cleave the DNA at these specific sites, generating fragments with defined terminal ends. Fragment selection: After the enzymatic digestion is complete, it is necessary to purify the products, select DNA fragments within a specific length range, ensure sequencing efficiency and data quality, and remove unreacted enzymes and other impurities. Common purification methods include:
Automated fragment recovery systems: Such as the Pippin Prep from Sage Science, which can precisely select DNA fragments within a specific length range (typically 300–500 bp). Agarose gel electrophoresis: DNA fragments of different lengths are separated by electrophoresis, and then the target gel bands of specific lengths are cut and the target fragments are recovered. The fragment selection and purification steps in ddRAD-seq are crucial for ensuring sequencing quality, and these steps can improve sequencing efficiency and the accuracy of the results.
Efficient Implementation of GBS Technology
Role of Restriction Enzymes: A single restriction enzyme (e.g., ApeKI) is used to cut genomic DNA, producing fragments of specific lengths. The enzyme is selected based on the distribution of its recognition sites in the target genome (e.g., frequency and uniformity) to ensure the fragments are suitable for sequencing.
Simplified Library Construction: After cutting, sequencing adapters are ligated to both ends of the fragments, which are then amplified by PCR and directly sequenced without the need for fragment selection, significantly simplifying the workflow.
Accurate control of 2b RAD in genome tagging
Unique Type of Restriction Enzymes: Utilizes type IIB restriction endonucleases such as BsaXI, AlfⅠ, and BaeⅠ. These enzymes are distinctive in that their recognition sites are located in the middle of the DNA double helix, yet they cleave on both the left and right sides of the recognition site, differing fundamentally from the cutting patterns of conventional restriction enzymes.
Uniform Length of Digested Products: The cleavage generates DNA fragments of fixed lengths, typically producing double-stranded DNA fragments (TAG tags) of uniform size, approximately 33-36 base pairs (bp). This consistency arises because type IIB restriction endonucleases excise DNA fragments of predetermined lengths from the genome. For example, BsaXI yields fragments of relatively fixed lengths, facilitating easier recovery and subsequent processing.
The flexibility and application expansion of ezRAD technology
Enzyme-free digestion: DNA is physically fragmented, followed by adapter ligation and sequencing.ezRAD technology enables genome fragmentation without relying on restriction enzymes, circumventing potential issues associated with enzymatic digestion, such as reduced efficiency due to genomic methylation modifications, the need for selecting specific enzymes, and the cost of purchasing multiple enzymes and adapters. This significantly lowers experimental complexity and costs. In addition to enzymatic digestion, ezRAD can utilize physical fragmentation methods, such as ultrasonication or high-pressure homogenization, to randomly shear DNA into fragments of desired lengths. While this approach avoids the specificity limitations of enzymatic digestion and enables unbiased genome coverage, it may compromise uniformity in fragment sizes. Alternatively, chemical methods—like inducing DNA strand breaks under extreme pH conditions—can also fragment genomic DNA. However, this requires strict control of reaction parameters to prevent excessive degradation. Overall, ezRAD enhances experimental flexibility by allowing researchers to choose the optimal fragmentation method based on laboratory conditions and study requirements, thereby broadening its applicability across diverse genomic studies.
How to Choose Different RAD-seq Technologies
Research Purpose: The required number of molecular markers varies depending on the research objectives. For instance, GWAS may demand tens of thousands of high-density molecular markers, whereas studies on phylogenetic relationships or linkage analysis typically require lower marker densities, often needing only hundreds to thousands of markers for successful analysis. Generally, the marker count follows the order: ddRAD ≥ ezRAD ≈ RAD > GBS > 2b-RAD. Therefore, selecting an appropriate sequencing technology should begin with evaluating the required number of markers for the study.
Reference Genome Availability: Does the study species have a reference genome? What is the quality of the reference genome assembly? If a reference genome is available: It allows for inferring missing genotypes and theoretically assessing enzyme - cutting site numbers. If there's no reference genome or it's of poor quality: ddRAD may be used more. It can perform local assembly to obtain fragments up to 400 - 500bp, which is good for SSR marker development and primer design. GBS can cluster reads to form consensus sequences and detect SNPs without a reference. However, 2b - RAD, with short fragments prone to repeat - sequence interference and less suitable for primer - based SNP validation, usually requires a reference genome.
Restriction Enzyme Selection: Determined by marker density requirements. The chosen enzyme should suit the species, considering factors like the number of repeats in the species where the enzyme acts. An enzyme suitable for one species may not be for another. Restriction fragments often have sticky ends, which may require designing different adapters.
DNA Sample Preparation: DNA sample quality and quantity: High - quality DNA is crucial for enzyme digestion, adapter ligation, and amplification. Also, different reduced - representation sequencing methods may have varying DNA quantity requirements.
Cost: GBS and 2b - RAD are relatively low - cost. GBS has simple library preparation and low sequencing costs, making it suitable for large - scale genetic diversity analysis. 2b - RAD has a significant cost advantage in high - density SNP development and is used for high - precision genetic mapping. ddRAD is of moderate cost and is used for studies with medium - sized samples to obtain medium - density SNPs. ezRAD, using patented reagent kits, has high library preparation and sequencing costs, but its simplicity and speed make it suitable for projects with moderate sample sizes and high time - cost sensitivity.
Technical Application: GBS is widely used for large - scale genetic diversity analysis and genome - wide association studies due to its simple library preparation and low cost. ddRAD is used for genetic diversity studies and QTL mapping in medium - sized samples, especially in complex genomes and non - model organisms. 2b - RAD, with its high - density SNP genotyping advantage, is used for high - precision genetic mapping and genetic analysis of important traits. ezRAD, with its simple operation, is suitable for projects with moderate sample sizes and high time - cost sensitivity.
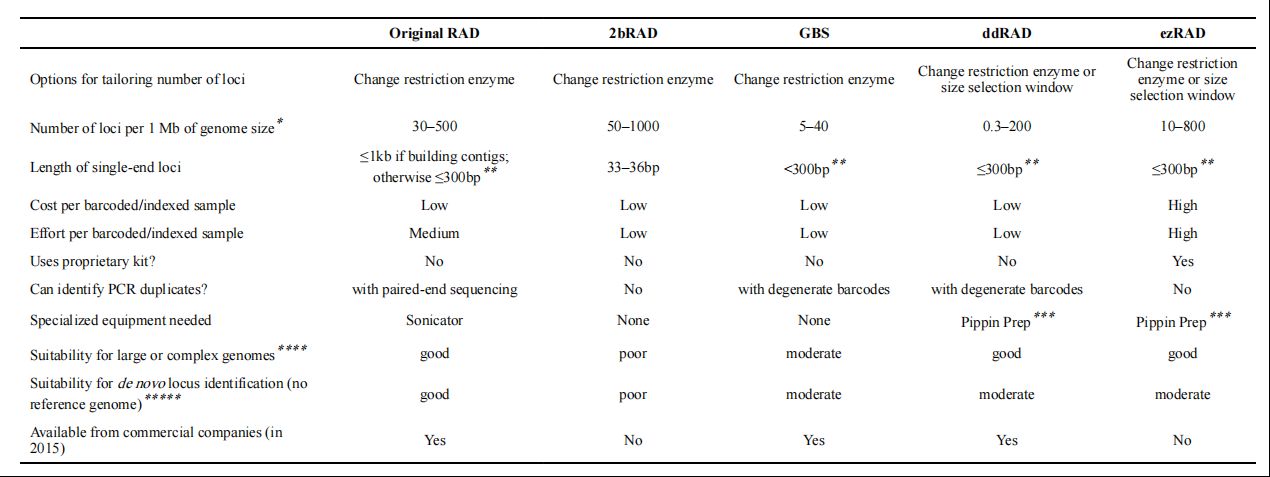 Summary of trade-offs among five RADseq methods (Kimberly R et al.,2016)
Summary of trade-offs among five RADseq methods (Kimberly R et al.,2016)
Applications of Different RAD-seq Technologies
RAD sequencing technologies are widely used inmolecular marker development, population genetics, genetic map construction, QTL mapping, GWAS, and molecular breeding. Taking the frequently cited GBS technology as an example, here are some applications of reduced-representation sequencing: For instance, a study utilized GBS and a recombinant inbred lines (RILs) mapping population to construct a high-density genetic map in maize, aiming to localize traits related to root architecture. Based on this map, 24 root structure-related traits were analyzed, identifying 62 QTL locithat accounted for 1.6%–11.6% of phenotypic variation. The study also revealed QTL clusters, suggesting these regions might harbor aggregated genes or pleiotropic loci associated with root architecture. This work laid the foundation for the precise identification of genes governing maize root structure.
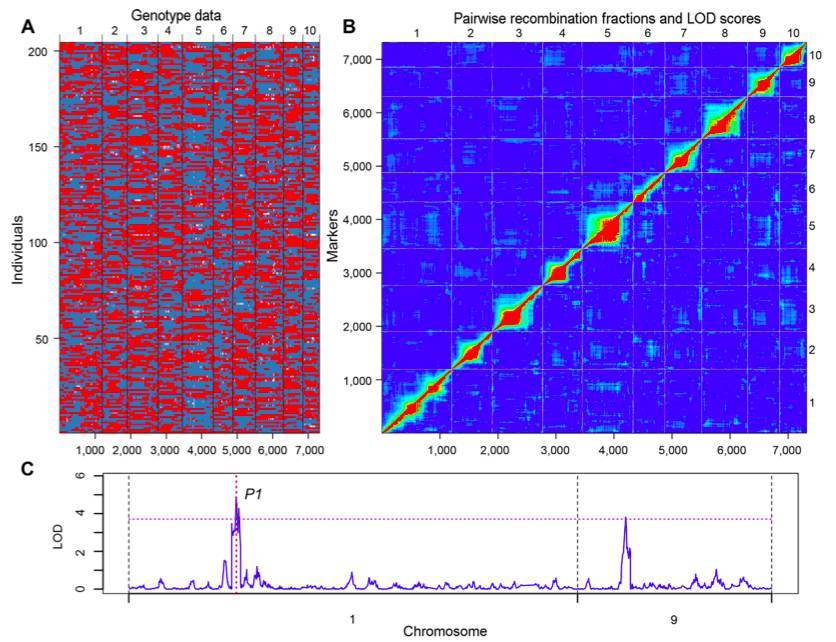 The results of bin map mapping of the RILs population and QTL mapping of the corn silk color gene (Song W et al.,2016)
The results of bin map mapping of the RILs population and QTL mapping of the corn silk color gene (Song W et al.,2016)
Some studies have conducted association analyses on alfalfa resistance to Verticillium wilt based on GBS technology. They performed genome - wide association analyses on alfalfa's resistance to Verticillium wilt using the GBS - based reduced - representation sequencing technology. Through association analysis, 10 SNPs significantly related to the target trait were identified. When comparing these 10 SNP markers with previous QTL results, it was found that the SNP markers on chromosome 7 and 8 were consistent with the previous QTL mapping results. The flanking sequences of the SNPs were compared using BLASTN to obtain candidate genes related to the trait. The development of markers related to Verticillium wilt resistance will aid in breeding resistant varieties.
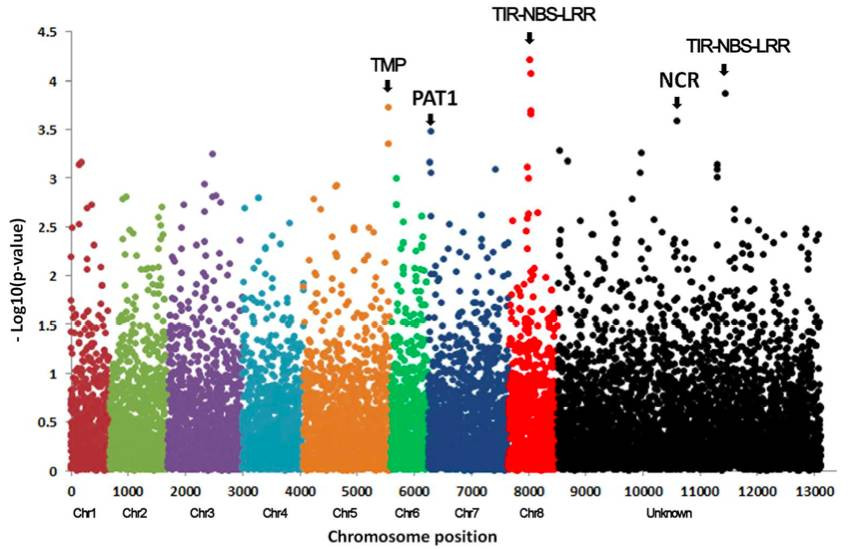 Results of genome-wide association analysis (Long-Xi et al.,2016)
Results of genome-wide association analysis (Long-Xi et al.,2016)
The ddRAD-seq technology is widely applied in genetic diversity analysis, population genetics studies, gene mapping, and kinship analysis, providing deep insights into the genetic structure of germplasm resources. Scholars have investigated genetic differentiation in Siberian larch (Larix sibirica) populations related to their adaptation to climatic conditions along altitudinal gradients. This research combined six bioclimatic variables with double-digest ddRAD-seq data to analyze population structure, genetic diversity, and genetic adaptation characteristics of Siberian larch to growth conditions within the Altai-Sayan mountain system. For the first time, the study revealed genetic differentiation in Siberian larch populations associated with adaptation to altitudinal climatic gradients. This was achieved through a joint analysis of elevation and six other bioclimatic variables, alongside a large set of genetic markers—SNPs—obtained from ddRAD-seq. A total of 25,143 SNPs were genotyped across 231 trees. Additionally, by selecting SNPs located outside the coding regions of the Siberian larch genome and mapping them to distinct fragments, a dataset of 761 putatively neutral SNPs was assembled. Using four analytical methods (PCAdapt, LFMM, BayeScEnv, and RDA), 550 outlier SNPs were identified. Among these, 207 SNPs exhibited significant correlations with environmental factors and were potentially linked to local adaptation. Notably, 67 SNPs were associated with elevation (identified via LFMM or BayeScEnv), with 23 SNPs supported by both methods.
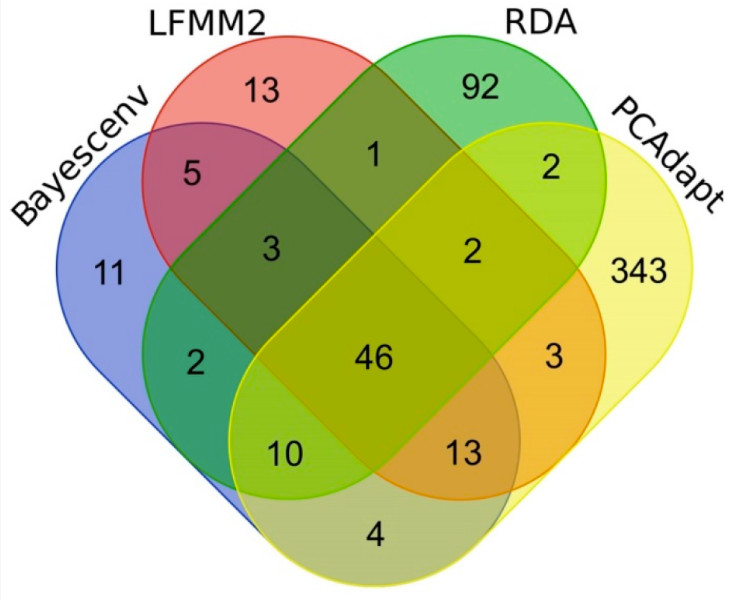 Venn diagram summarizing the results of the search for significant outlier SNPs (Serafima V et al.,2023)
Venn diagram summarizing the results of the search for significant outlier SNPs (Serafima V et al.,2023)
In a study on soybean photoperiodic flowering and regional adaptation, the 2b-RAD technology was employed to construct a high-density genetic map of a recombinant inbred line (RIL) population derived from the cross between the extremely late-maturing cultivar "Zigongdongdou" and the extremely early-maturing cultivar "Heihe 27." This enabled the identification of the major QTL qFT12-2, which governs flowering time and was further resolved as the circadian clock gene GmPRR37. By integrating phenotypic data from multiple environments and population genetic analyses, the study revealed that natural variations in GmPRR37 (e.g., a nonsense mutation in early-maturing cultivars leading to the loss of the CCT domain) and its diurnal expression patterns critically regulate photoperiod adaptation. CRISPR/Cas9-mediated gene-editing experiments confirmed that loss-of-function mutations in GmPRR37 significantly advanced flowering under long-day conditions (by 15.8 days), while overexpression delayed flowering. The 2b-RAD technology provided critical molecular marker support for deciphering the genetic effects of GmPRR37, its natural selection mechanisms, and its central role in soybean latitudinal adaptation.
 High-density linkage maps of 20 linkage groups (Liwei et al.,2020)
High-density linkage maps of 20 linkage groups (Liwei et al.,2020)
Some scholars have experimentally verified the effectiveness and feasibility of ezRAD-seq technology in genomic genotyping of non - model organisms, and explored its potential applications in different research fields such as ecology, evolutionary biology, and conservation biology.
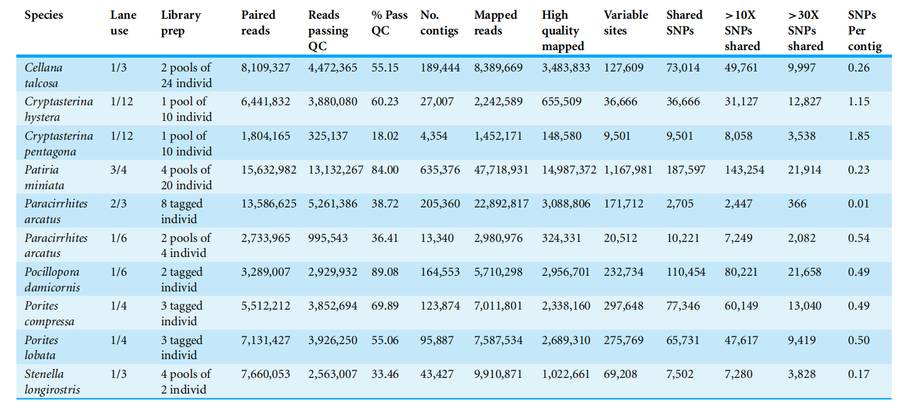 Summary of ezRAD results of the two channels sequenced by Illumina GAIIx (Robert J et al., 2013)
Summary of ezRAD results of the two channels sequenced by Illumina GAIIx (Robert J et al., 2013)
Summary
RAD-seq is a high-throughput sequencing technology that uses restriction enzyme sites for genomic sequencing. It can obtain high-density SNP data from numerous individuals and is widely used in fields like evolutionary biology and ecology. Since its introduction in 2008, several variants, including Original RAD-seq, GBS, ddRAD-seq, 2bRAD, and ezRAD, have been developed. They vary in digestion methods, fragment selection criteria, and applications. Choosing the appropriate technology depends on research objectives, reference genome availability, enzyme selection, DNA sample preparation, and cost. These technologies are crucial for molecular marker development, population genetics, genetic mapping, QTL identification, genome-wide association studies, and molecular breeding, serving as powerful tools in genomics research.
References
- Baird, N. A., et al. "Rapid SNP Discovery and Genetic Mapping Using Sequenced RAD Markers." PLoS ONE.2008, 3(10):e3376 https://doi.org/10.1371/journal.pone.0003376
- Kimberly R. AndrewsCA, Jeffrey M. Good, Michael R. Miller, et al. "Harnessing the power of RADseq for ecological and evolutionary genomics" Nature Reviews Genetics,2016, 17 (2): 81-92 https://doi.org/10.1038/nrg.2015.28
- Song W, Wang B, Hauck A L, et al. "Geneticdissection of maize seedling root system architecture traits using anultra-high density bin-map and a recombinant inbred line population" Journal of Integrative Plant Biology.2016, 58(3) https://doi.org/10.1111/jipb.12452
- Long-Xi, Yu, Ping, Zheng, Tiejun, Zhang, et al. "Genotyping-by-sequencing-based genome-wide association studies on Verticillium wilt resistance in autotetraploid alfalfa (Medicago sativa L.)" Mol Plant Pathol. 2016, 18: 0 https://doi.org/10.1111/mpp.12389
- Serafima V, Novikova, Vadim V, Sharov, et al. "Genetic Adaptation of Siberian Larch (Larix sibirica Ledeb.) to High Altitudes" Int J Mol Sci. 2023, 24: 0 https://doi.org/10.3390/ijms24054530
- Liwei, Wang, Shi, Sun, Tingting, et al. "Natural variation and CRISPR/Cas9-mediated mutation in GmPRR37 affect photoperiodic flowering and contribute to regional adaptation of soybean" Plant Biotechnol J. 2020, 18: 0 https://doi.org/10.1111/pbi.13346
- Robert J, Toonen, Jonathan B, Puritz, Zac H, "et al. "ezRAD: a simplified method for genomic genotyping in non-model organisms" PeerJ. 2013, 1: 0 https://doi.org/10.7717/peerj.203


 Send a Message
Send a MessageFor any general inquiries, please fill out the form below.