
ddRAD-seq Explained: Definition and Principle, Key Applications and Future Trends in Agriculture
With the development of genome sequencing technology, ddRAD-seq (double digestion restriction fragment dimensionality reduction sequencing) has become widely popular in the field of molecular ecology due to its use of high-throughput sequencing (NGS) platforms to develop novel SNP (single nucleotide polymorphism) markers. ddRAD-seq is a double enzyme digestion simplified genome sequencing technology developed on the basis of traditional RAD-seq. The main difference between ddRAD-seq and RAD-seq is that genomic DNA is double digested by combining a rare restriction endonuclease with a common restriction endonuclease, eliminating the step of interruption and directly screening fragments for sequencing. Meanwhile, this method utilizes the site specificity of restriction endonucleases to generate library fragments from conserved genomic regions in different individuals of the same species. This conservatism enables sequencing and comparative analysis of the same genomic regions among different individuals. Therefore, ddRAD-seq can quickly and efficiently develop a large number of genetic markers.
This article comprehensively reviews the technical principles, advantages, limitations, and applications of ddRAD-seq technology. At the same time, the important applications of ddRAD-seq technology in agricultural genomics research and molecular breeding were analyzed. With its high throughput, low cost, and high resolution, ddRAD-seq will further promote crop improvement and sustainable agricultural development.
What is ddRAD-seq
ddRAD-seq is a simplified genome sequencing technology based on double enzyme digestion, mainly used for discovering single nucleotide polymorphisms (SNPs) and genotyping. It is a powerful tool widely used in ecology, evolutionary biology, and genetics research, especially with significant value in the study of non model organisms.
Principle Overview of ddRAD-seq
ddRAD-seq is a reduced-representation genome sequencing technique based on double enzyme digestion, primarily used for single nucleotide polymorphism (SNP) discovery and genotyping. Its principle involves reducing genome complexity through double digestion and fragment size selection to enable sequencing of specific genomic regions. The core steps of ddRAD-seq are as follows:
Genomic DNA Extraction: High-quality genomic DNA is isolated from tissue samples.
Double Digestion Reaction: Genomic DNA is digested with two restriction enzymes to produce restriction fragments. One enzyme typically recognizes rare restriction sites while the other targets more frequent sites, generating fragments of specific lengths through dual digestion.
Ligation Reaction: T4 DNA ligase is used to attach barcoded adapters to the digested fragments. These adapters contain sample-specific barcode sequences for multiplexing different samples.
Fragment Size Selection: DNA fragments within a specific size range are selected using agarose gel electrophoresis or automated size selection systems (e.g., Pippin Prep). This step ensures retention of appropriately sized fragments for downstream analysis.
PCR Amplification: Selected DNA fragments undergo PCR amplification to increase template quantity and enrich target fragments. Sequencing platform-specific primer sequences are simultaneously incorporated during PCR.
Sequencing: Purified DNA fragments are subjected to high-throughput sequencing to obtain simplified genomic data.
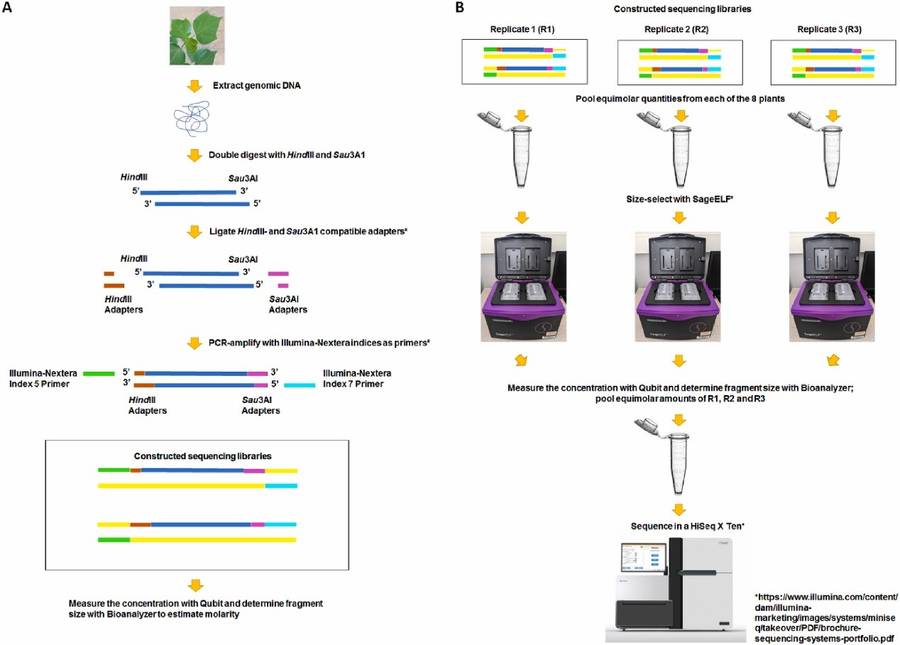 Flowchart of the ddRAD-seq methodology (Magbanua et al.,2023)
Flowchart of the ddRAD-seq methodology (Magbanua et al.,2023)
Technical Advantages and Limitations of ddRAD-seq
ddRAD-seq has emerged as an ideal choice due to its technical advantages in cost-effectiveness, high efficiency, flexibility, and resolution, particularly suited for large-scale population genetics studies and genomic analyses of non-model organisms. In plant and agricultural research, the ddRAD-seq technology demonstrates numerous advantages but also has some limitations.
Advantages of ddRAD-seq
Cost-Effectiveness and High Efficiency: By combining double enzyme digestion and fragment size selection to target DNA fragments of specific lengths, ddRAD-seq significantly reduces the genomic regions requiring sequencing. This approach minimizes sequencing data volume, enhances sequencing efficiency, and lowers costs. Its reference genome-independent workflow makes it especially advantageous for non-model organisms or species with incomplete genomic resources, further reducing experimental expenses. Through barcode design, ddRAD-seq enables multiplexing of multiple samples for high-throughput analysis, making it ideal for large-scale population genetics studies while enhancing experimental efficiency.
High Flexibility: ddRAD-seq allows researchers to customize restriction enzyme combinations to adjust genome complexity and fragment lengths based on study objectives. This adaptability ensures compatibility with diverse research goals and species-specific genomic characteristics.
High Resolution: ddRAD-seq generates high-density single nucleotide polymorphism (SNP) datasets with exceptional accuracy and reproducibility, enabling high-resolution genetic analyses. Its compatibility with high-throughput sequencing platforms (e.g., Illumina) ensures the production of high-quality sequencing data, further enhancing resolution capabilities.
Limitations of ddRAD-seq
Uneven Marker Coverage: Due to ddRAD-seq's reliance on specific restriction enzyme recognition sites, marker distribution may be influenced by the genomic distribution of these enzyme cleavage sites, leading to uneven coverage. Some regions may have abundant markers, while others may have sparse or no markers, potentially hindering the study and analysis of certain genomic regions.
High Experimental Complexity: The ddRAD-seq workflow is relatively intricate, involving multiple steps such as genomic DNA extraction, enzymatic digestion, adapter ligation, fragment selection, and PCR amplification. These steps require precise execution and optimization to ensure experimental success and data quality. Any errors or deviations during the process may result in inaccurate or unreliable data.
Complex Bioinformatics Analysis: The large datasets generated by ddRAD-seq demand sophisticated bioinformatics processing, including data filtering, alignment, SNP calling, and analysis. This necessitates specialized bioinformatics expertise, advanced computational resources, and may pose challenges for researchers lacking relevant skills or infrastructure.
Genotype Missingness: During ddRAD-seq, genotype missingness may occur due to incomplete enzymatic digestion, low adapter ligation efficiency, fragment loss, or insufficient sequencing depth. Such missing data can compromise the completeness and accuracy of results, impacting downstream analyses and research conclusions.
Impact of Restriction Site Polymorphisms: If polymorphisms occur precisely at restriction enzyme recognition sites, they may cause allele dropout or null alleles. This can reduce genotyping accuracy and introduce biases in genetic diversity assessments, gene mapping, and related studies.
Services you may interested in
How Does ddRAD-seq Empower Agriculture
In the agricultural field, ddRAD-seq technology demonstrates tremendous application potential, providing robust support for plant genetic diversity research, genetic map construction, agronomic trait gene mapping, and plant breeding. Through this technology, agricultural researchers can deeply explore the genetic potential of crops and promote the sustainable development of modern agriculture.
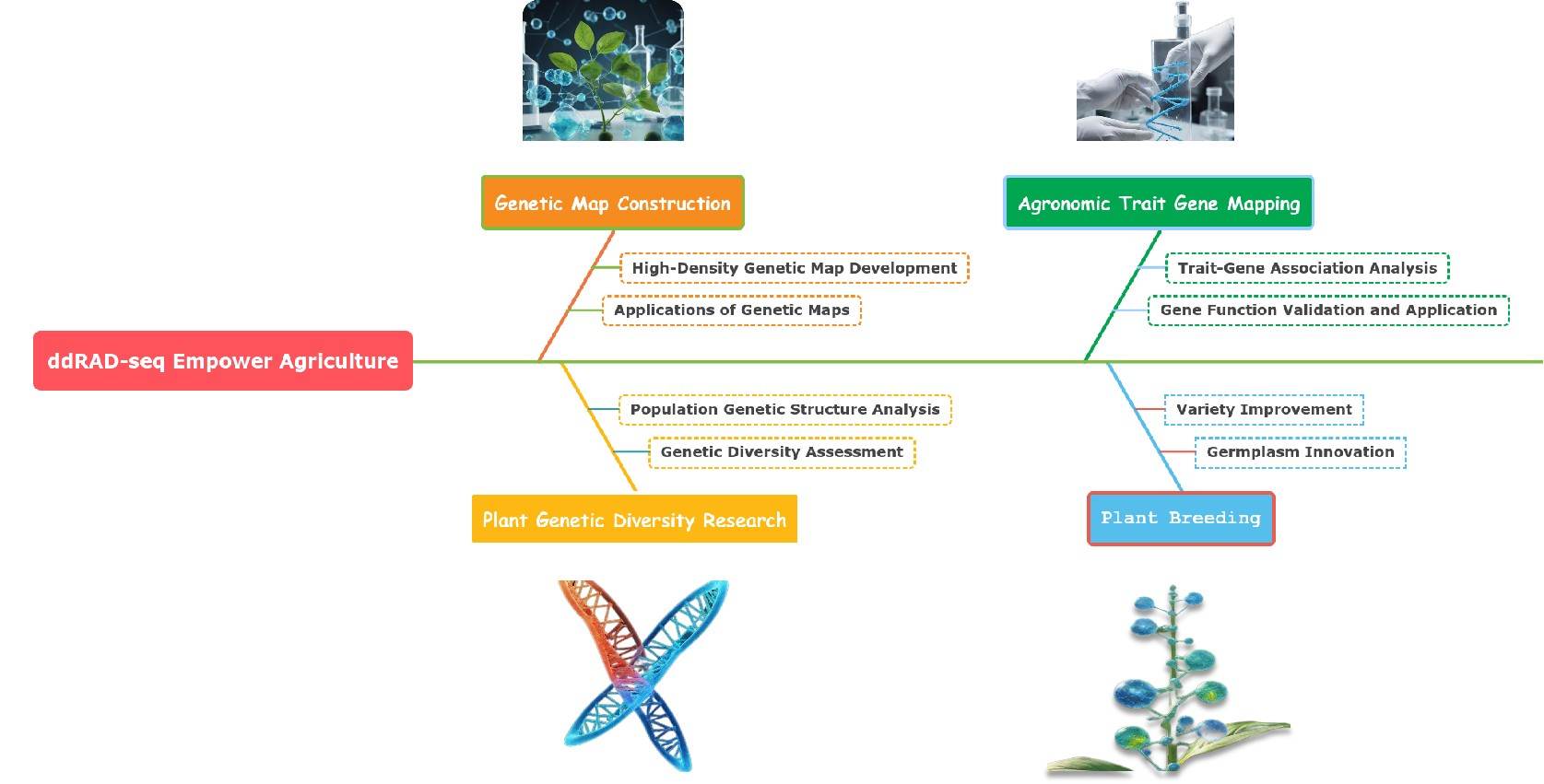 Key Applications of ddRAD-seq in Agriculture.
Key Applications of ddRAD-seq in Agriculture.
Plant Genetic Diversity Research
Population Genetic Structure Analysis: ddRAD-seq enables genetic structure analysis of plant populations, revealing genetic differentiation and adaptive evolutionary mechanisms among different populations. For example, in crops such as tomatoes and wheat, ddRAD-seq has been used to study the genetic structures of local varieties and wild species, providing scientific guidance for the conservation and management of agricultural germplasm resources.
Genetic Diversity Assessment: ddRAD-seq efficiently evaluates the genetic diversity of plant germplasm resources. By analyzing the distribution and frequency of SNP markers, researchers gain deeper insights into the genetic potential of germplasm resources, offering valuable genetic materials for breeding new varieties.
A research team at Fudan University developed a universal computational tool to optimize plant RAD-seq design using ddRAD-seq technology, with experimental validation demonstrating its effectiveness. The optimized RAD-seq approach achieved chloroplast DNA and rRNA gene removal during library construction while maintaining desired coverage uniformity and density. Through strategic restriction enzyme (RE) selection optimization and implementation of precise DNA fragment size-selection technology, this method significantly enhanced RAD-seq efficiency and accuracy. The Pippin Prep system was utilized as the DNA fragment size-selection tool at two critical stages of library preparation. Post-selection, over 90% of sequenced fragments in each library fell within the target size range (220-420 bp).
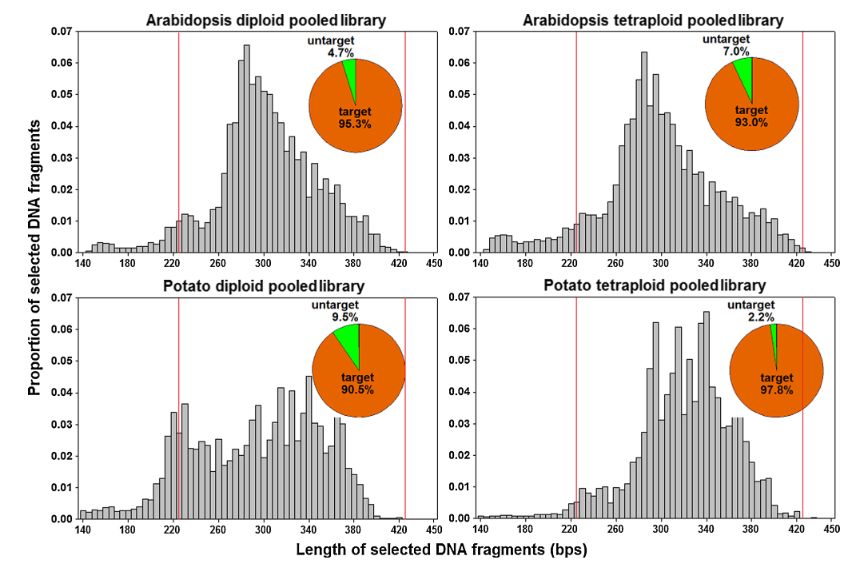 Length distribution of sequenced DNA fragments in each pooled RAD-seq dataset (Jiang et al.,2016)
Length distribution of sequenced DNA fragments in each pooled RAD-seq dataset (Jiang et al.,2016)
Genetic Map Construction
High-Density Genetic Map Development: ddRAD-seq rapidly generates large numbers of SNP markers to construct high-density genetic maps. Through the application of ddRAD-seq technology, researchers successfully constructed a high-density genetic map for oil-tea camellia spanning 3,327 cM. This map comprises 2,780 molecular markers distributed across 15 linkage groups, with an average marker interval of 1.20 cM. The technology further enabled the identification of 221 quantitative trait loci (QTLs) associated with fruit and oil-related traits through three-year phenotypic data analysis, among which 17 major QTLs accounted for 13.0% to 16.6% of phenotypic variation. Collinearity analysis between the genetic map and the "CON" genome of oil-tea camellia revealed 202 potential candidate genes within QTL regions, functionally linked to fruit development and lipid biosynthesis. Notably, this study demonstrates the significant utility of ddRAD-seq technology in molecular breeding of oil-tea camellia. By identifying trait-associated QTLs and adjacent SNP markers, researchers have developed molecular markers for marker-assisted selection (MAS), providing crucial tools to accelerate the genetic improvement of this economically important species.
 ddRAD based high-density genetic map of 180 full-sibs (Lin et al.,2024)
ddRAD based high-density genetic map of 180 full-sibs (Lin et al.,2024)
Applications of Genetic Maps: High-density genetic maps play critical roles in gene mapping, quantitative trait locus (QTL) analysis, and marker-assisted breeding. These maps allow precise localization of genes associated with key agronomic traits, accelerating breeding processes.
Agronomic Trait Gene Mapping
Trait-Gene Association Analysis: ddRAD-seq identifies genes linked to important agronomic traits. For example, in mustard (Brassica juncea), ddRAD-seq-based association analysis successfully identified SNP markers associated with multiple agronomic traits.
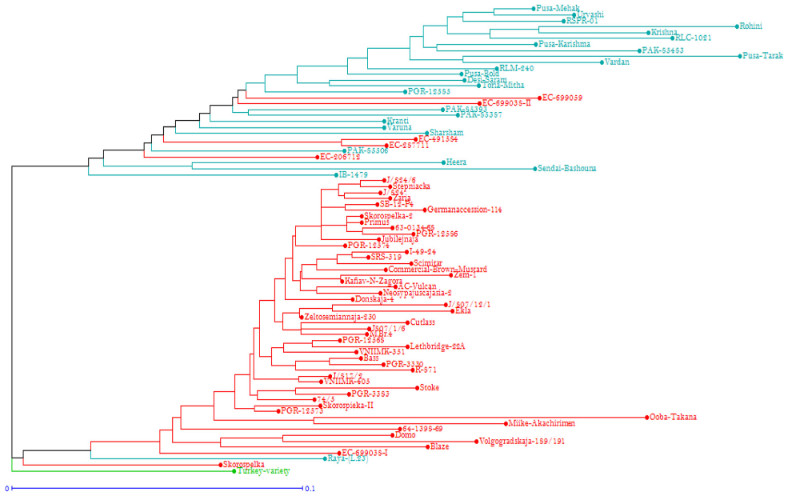 Genetic relationship among 80 different Brassica juncea genotypes (Sudan et al.,2019)
Genetic relationship among 80 different Brassica juncea genotypes (Sudan et al.,2019)
Gene Function Validation and Application: The Italian Research Centre for Vegetable and Ornamental Crops used ddRAD-seq technology to analyze the genetic diversity of 288 tomato samples. This included 152 samples of the long shelf life (LSL) germplasm variety 'da serbo', which mainly originated from Italy and Spain, with the remaining samples from common varieties in other countries. In addition to its LSL trait, the 'da serbo' variety also exhibited stress resistance. The study identified 32,799 high-quality SNPs, which were used for ancestry population structure simulation and non-parametric hierarchical clustering. The research delineated six genetic subgroups, clearly separating most of the 'da serbo' varieties and indicating population stratification based on type and geographic origin. Linkage disequilibrium (LD) decayed rapidly within a range of less than 5 kb. By analyzing SNPs with minor allele frequency (MAF) in the 'da serbo' variety, the authors found that the stress tolerance of this germplasm was associated with high-frequency mutations in genes related to fruit ripening, such as CTR1 and JAR1. Finally, 58 core materials containing most of the diversity were selected to further develop key traits. This study shows that the 'da serbo' germplasm selected in the Mediterranean Basin has genetic imprints. Additionally, the study provides new insights into the LSL 'da serbo' germplasm as a source of stress-resistant alleles.
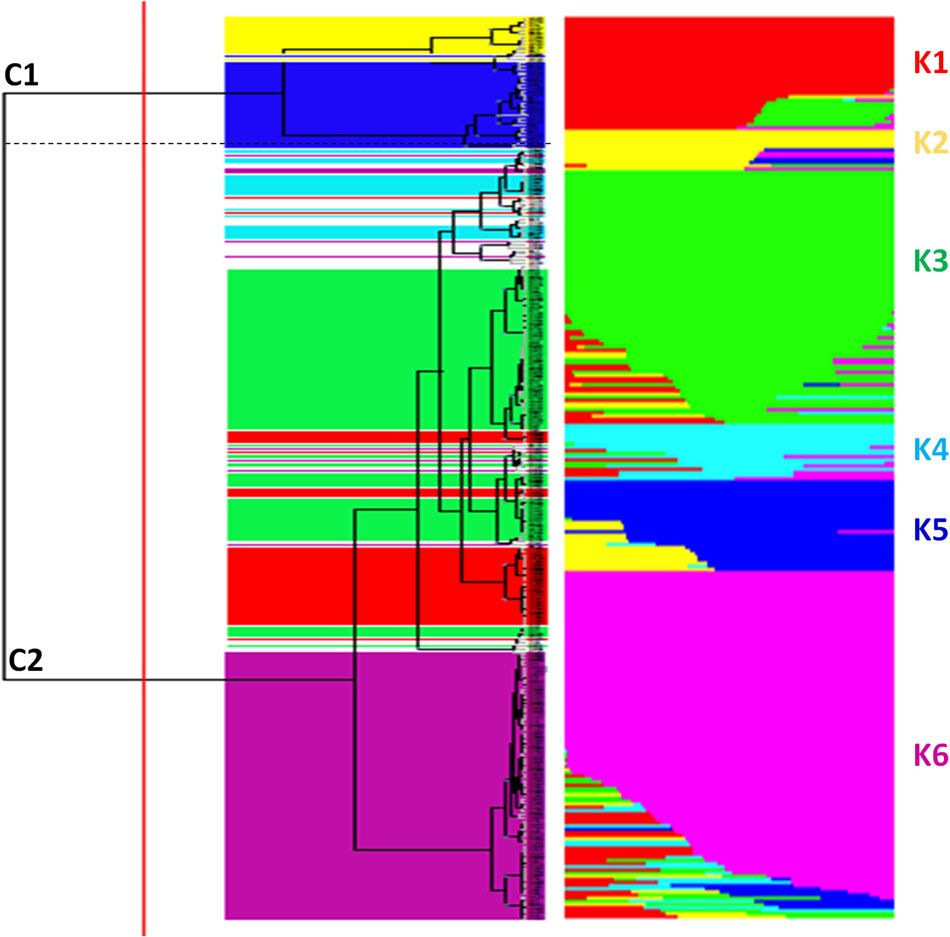 Estimate of genetic diversity in 288 tomato accessionsusing ddRAD (Esposito et al.,2020)
Estimate of genetic diversity in 288 tomato accessionsusing ddRAD (Esposito et al.,2020)
Plant Breeding
Variety Improvement: The ddRAD-seq technology provides unprecedented resolution for crop breeding through genome-wide high-density single nucleotide polymorphism (SNP) marker analysis. In maize (Zea mays), researchers utilized drought resistance and grain yield co-localized quantitative trait loci (QTLs) identified by ddRAD-seq to successfully develop new cultivars that reduce yield loss by 35% under drought conditions. Studies further demonstrate that integrating ddRAD-seq data with machine learning algorithms enables the prediction of phenotypic values in hybrid progenies, increasing the screening accuracy of superior genotypes to 92% and significantly reducing the scale of field trials.
Germplasm Innovation: The synergistic application of ddRAD-seq and genome editing tools such as CRISPR-Cas9 bridges the gap from "natural variation mining" to "artificial directional design." For instance, pan-genome analysis of soybean (Glycine max) germplasm resources via ddRAD-seq revealed rare SNPs regulating photoperiod sensitivity (e.g., variations in the GmFT2a promoter region). These SNPs were precisely introduced into commercial cultivars using a cytosine base editor (CBE), successfully overcoming latitudinal adaptability constraints. In woody plants, the targeted capture capability of ddRAD-seq efficiently identifies low-frequency recombination events, providing high-confidence targets for genome editing. For example, in poplar (Populus spp.), SNPs in the lignin biosynthesis pathway identified by ddRAD-seq guided multiplex CRISPR editing to simultaneously knock out the CCoAOMT and COMT genes, resulting in engineered germplasm with an 18% increase in cellulose content and unaffected growth rates. Notably, ddRAD-seq facilitates cross-species gene resource utilization: haplotype analysis of wild rice (Oryza rufipogon) via ddRAD-seq identified novel haplotypes in the Pi9 gene cluster conferring blast resistance. After editing and introgression into cultivated rice (Oryza sativa), disease incidence decreased by 67%.
Future Perspectives of ddRAD-seq
Technological Improvements
Enhancing Sequencing Throughput and Accuracy: With continuous advancements in sequencing technology, future ddRAD-seq protocols are expected to achieve higher sequencing throughput, enabling the analysis of more samples in shorter timeframes. Concurrently, optimizing sequencing workflows and data processing algorithms will improve accuracy, reducing sequencing errors and genotype missingness.
Reducing Experimental Costs: Current ddRAD-seq workflows involve relatively high costs, particularly in library preparation and sequencing. Future efforts could focus on developing cost-effective reagents, streamlining experimental protocols, and increasing automation to lower expenses, making the technology more accessible for large-scale agricultural research and breeding programs.
Streamlining Data Analysis: The massive datasets generated by ddRAD-seq require complex bioinformatics analysis. Developing user-friendly, efficient bioinformatics tools and software will simplify data processing, reduce reliance on specialized expertise, and empower more researchers to analyze and interpret results independently.
Improving Marker Coverage Uniformity: To address uneven marker distribution caused by reliance on restriction enzyme sites, future improvements may include optimizing enzyme selection strategies, refining library construction methods, and integrating complementary sequencing technologies. This would enhance genome-wide coverage and provide a more comprehensive view of genetic variation.
Minimizing Genotype Missingness: Genotype missingness, a critical factor affecting data quality, can be mitigated by optimizing experimental conditions (e.g., enzyme digestion efficiency), increasing sequencing depth, and refining data processing algorithms. These measures will improve data completeness and reliability.
Application Prospects
Precision Agriculture: ddRAD-seq can elucidate genotype-phenotype relationships and crop adaptability to environmental conditions. Precise genotyping and genetic analysis will support precision agriculture by guiding farmers in selecting locally optimized crop varieties, refining cultivation practices, and enhancing agricultural productivity and sustainability.
Smart Breeding: Integrating ddRAD-seq with modern information technologies enables intelligent breeding management and decision-making. Large-scale genetic diversity analysis and gene mapping accelerate the identification of elite genetic combinations. Additionally, genomic prediction and selection powered by ddRAD-seq enhance breeding precision and efficiency, advancing smart breeding objectives.
Germplasm Conservation and Utilization: ddRAD-seq facilitates efficient genetic diversity assessments and structural analyses of plant germplasm, providing critical insights for conservation strategies. By uncovering valuable agronomic traits and genes, this technology enriches genetic resources for breeding novel varieties and promotes sustainable utilization of germplasm.
Integration with Emerging Technologies: Combining ddRAD-seq with genome editing enables precise genomic modifications to develop superior crop traits. Targeting and editing specific genes can expedite the creation of high-performance varieties. Furthermore, synergies with synthetic biology and bioinformatics offer innovative solutions for sustainable agricultural development.
References
- Magbanua Z V, Hsu C Y, Pechanova O, et al. "Innovations in double digest restriction-site associated DNA sequencing (ddRAD-Seq) method for more efficient SNP identification" Analytical Biochemistry. 2023, 662 https://doi.org/10.1016/j.ab.2022.115001
- Ning Jiang, Fengjun Zhang, Jinhua Wu, et al. "A highly robust and optimized sequence-based approach for genetic polymorphism discovery and genotyping in large plant populations" Theoretical and Applied Genetics. 2016 https://doi.org/10.1007/s00122-016-2736-9
- Lin P, Chai J, Wang A, et al."High-Density Genetic Map Construction and Quantitative Trait Locus Analysis of Fruit- and Oil-Related Traits in Camellia oleifera Based on Double Digest Restriction Site-Associated DNA Sequencing" International Journal of Molecular Sciences. 2024, 25(16) https://doi.org/10.3390/ijms25168840
- Sudan J, Singh R, Sharma S, et al."ddRAD sequencing-based identification of inter-genepool SNPs and association analysis in Brassica juncea" BMC Plant Biology. 2019, 19(1) https://doi.org/10.1186/s12870-019-2188-x
- Esposito S, Cardi T, Campanelli G, et al."ddRAD sequencing-based genotyping for population structure analysis in cultivated tomato provides new insights into the genomic diversity of Mediterranean'da serbo'type long shelf-life germplasm" Horticultural Research. 2020(001):007 https://doi.org/10.1038/s41438-020-00353-6


 Send a Message
Send a MessageFor any general inquiries, please fill out the form below.