
The Future of Precision Breeding: Integrating MAS With Genomic Selection for Complex Traits

A single, pragmatic question sits behind most modern breeding programs: how can a team move faster on complex traits without sacrificing reliability? For rice, one compelling answer is to integrate MAS and genomic selection in a single, governed workflow—using marker-assisted selection (MAS) to lock in validated blast resistance loci and genomic selection (GS) to rank for multi-environment yield stability. That division of labor is the core of hybrid pipelines that many teams now adopt under the banner of "precision breeding." In these pipelines, MAS provides clear go/no-go decisions at known loci, while GS captures the diffuse, genome-wide signal that drives background gain. Think of it this way: MAS is the safety gate; GS is the accelerator.
This guide distills the why and how—anchored on the rice use case (blast resistance + yield stability) and a deployment strategy that favors low-coverage whole-genome sequencing (LC-WGS) with imputation, complemented by a small panel of validated decision markers. It couples anonymized operational ranges with authoritative references so that program managers and bioinformatics leads can take these ideas from slideware to practice.
Key takeaways
- Hybrid pipelines are most effective when MAS secures validated, large-effect loci and GS drives rank gains on polygenic backgrounds. The two methods are complementary, not competitive.
- A simple boundary rule helps: if major, deployable loci exist, use MAS as a hard gate; when many small effects dominate, rely on GS for early ranking across environments. Hybridization shines when both conditions hold.
- Training population design and phenotype strategy determine most of GS accuracy; model choice matters far less than data quality and alignment to deployment use cases.
- At deployment scale, LC-WGS with imputation often balances density, flexibility, and cost—provided a representative reference panel exists—while a small, robust MAS panel de-risks decisions.
- Governance keeps hybrid programs from drifting: version marker sets and models, monitor batch effects and realized predictive ability, and define retraining triggers.
Why MAS and Genomic Selection Together Beat Either Alone for Many Programs
Hybrid strategies use MAS to lock in large-effect loci while GS captures the polygenic background that drives most complex-trait gain. In other words, MAS provides decisive checks at actionable genes, and GS leverages genome-wide markers to estimate breeding values earlier in the cycle. Multiple reviews converge on this logic: MAS remains efficient for major genes and targeted introgression, whereas GS excels for quantitative traits where many small effects aggregate into performance; together, they map neatly onto real trait architectures across crops. For a concise orientation to GS fundamentals, see the overview on genomic selection in plant and animal breeding. For a refresher on the MAS concept and use cases, see this marker-assisted selection (MAS) primer.
What MAS Is Best At (Actionable Loci, Clear Go/No-Go)
MAS is ideal when validated, large-effect loci drive material risk or benefit. Rice blast resistance genes such as Pi9, Pi54, Pigm, and others illustrate why: a small number of well-characterized loci can transform disease outcomes when reliably deployed. Studies have reported durable, broad-spectrum performance when genes like Pigm are introgressed into elite backgrounds with functional or tightly linked markers, with lines advancing to official release without yield penalties—see the 2022 report on Pigm deployment and variety approval for geng/japonica backgrounds in Jiangsu in Feng and colleagues' study. Pyramiding resistance loci (for example, Pi9 with Pi54) is a frequent MAS objective; marker-assisted backcrossing can secure double-homozygous plants while preserving agronomic traits, as summarized in Thulasinathan et al. (2023).
What GS Is Best At (Genome-Wide Signal, Early Ranking)
GS leverages genome-wide markers to predict breeding values for complex traits—like yield stability across environments—where hundreds or thousands of loci contribute small effects. The most consistent message in recent syntheses is that training population composition and multi-environment phenotyping dominate predictive ability, while the model choice itself is secondary. Reviews across crops indicate that aligning the training set to the target germplasm and deploying multi-environment or multi-trait multi-environment models can materially improve realized prediction, as summarized by Lee et al. (2024) and multi-environment case studies in wheat and sesame (e.g., Gill et al., 2021 and Sabag et al., 2023).
Where Hybrid Delivers the Biggest ROI (Cycle Time, Risk, and Scale)
Teams realize the most value from MAS and genomic selection combined when they can simultaneously reduce cycle time and de-risk decisions. For rice, MAS gates can enforce the presence of key blast resistance loci before lines proceed; GS then ranks the remaining candidates for yield stability across multi-environment trials. The net effect is fewer late-stage surprises—both disease and performance—while retaining throughput. Is there a universal, peer-reviewed, head-to-head comparison proving hybrid always wins? Not yet in 2020–2026 literature; however, logic supported by MAS field deployments and GS accuracy studies makes a strong operational case.
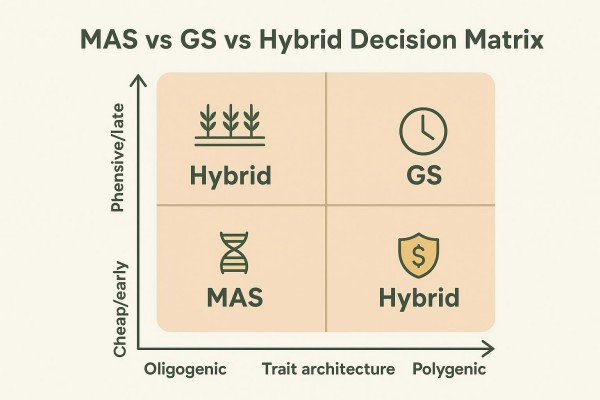
A Simple Boundary Rule for Complex Traits
A working rule helps teams decide how much to rely on MAS versus GS at each stage: MAS targets validated loci with deployable effect sizes; GS is the default when many small effects dominate prediction accuracy. Hybrid strategies are often a strong option when trait architecture mixes major loci and polygenic background, especially when phenotyping is expensive, late, or environment-sensitive.
For historical context on when MAS outperforms pure phenotypic selection and when it does not, this comparison of MAS versus phenotypic selection outlines boundaries and caveats.
"Major Locus vs Polygenic Background" in Practical Terms
In practice, a team defines "major" by the magnitude and stability of the effect, the availability of robust, deployable assays, and the absence of deleterious linkage drag in the target backgrounds. The "polygenic background" is everything else: the cumulative effect of many small alleles interacting with environments to drive yield stability, quality, and other complex outcomes.
What "Validated" Means Before a Locus Enters a Hybrid Pipeline
Validation means more than a significant p-value. Operationally, programs require cross-background assay robustness, replicated phenotypic effects across years/sites or independent validation panels, and clear, deployable effect sizes. As a reference point, rice disease-resistance programs have long progressed only when functional or tightly linked markers were available and confirmed across diverse panels, as reflected in Feng et al. (2022) and Thulasinathan et al. (2023).
The Most Common Misclassification Errors (and How to Avoid Them)
Two mistakes recur: first, treating medium-effect loci as "major" without sufficient cross-background checks, which produces brittle MAS gates; second, underestimating G×E and over-fitting models trained on poorly aligned or under-phenotyped datasets. Both failures are avoidable with stricter validation criteria for decision markers and with training populations deliberately aligned to deployment germplasm and environments.
Two Proven Hybrid Playbooks
Most real-world integrations look like one of two patterns, chosen according to risk tolerance and throughput needs.
Playbook A — Fix Major Loci First, Then Run GS for Background Gain
Here, MAS acts as a hard prefilter for validated resistance loci (e.g., Pi9, Pi54, or Pigm). Candidates that pass the MAS gate then enter GS ranking for yield stability and related quantitative traits across environments. This approach de-risks disease exposure early and is especially attractive when the MAS assays are robust and the resistance portfolio benefits from pyramiding—an approach consistent with Thulasinathan et al., 2023 and Feng et al., 2022.
Playbook B — Run GS First, Then Apply MAS as a Constraint or Safety Gate
When early throughput rules, teams may first run LC-WGS with imputation and compute GEBVs to rank large cohorts, then apply the MAS gate near the advancement decision. This playbook preserves scale for discovering polygenic winners while keeping disease risk in check at the point of action.
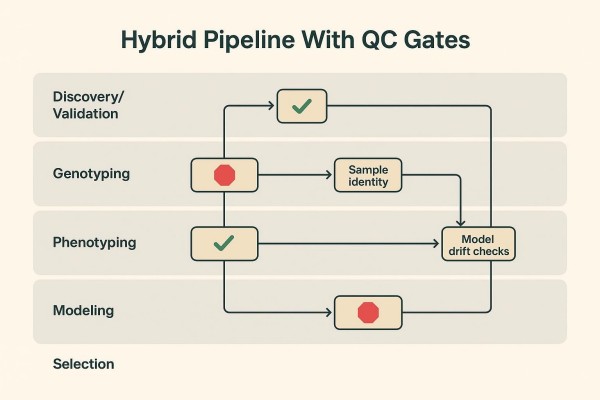
Where QC Gates Belong (So "Hybrid" Doesn't Become "Hybrid Chaos")
Quality gates should be explicit and auditable. Typical gates include: marker robustness verification for decision assays; sample identity checks and contamination/duplicate detection; batch comparability thresholds for genome-wide marker sets; model drift monitors tied to realized predictive ability; and documented acceptance criteria before selections advance. General QC frameworks from the genotyping literature recommend controlling missingness, call rates, and population structure signals (e.g., PCA by batch) to avoid spurious inferences, as outlined by Pavan et al. (2020).
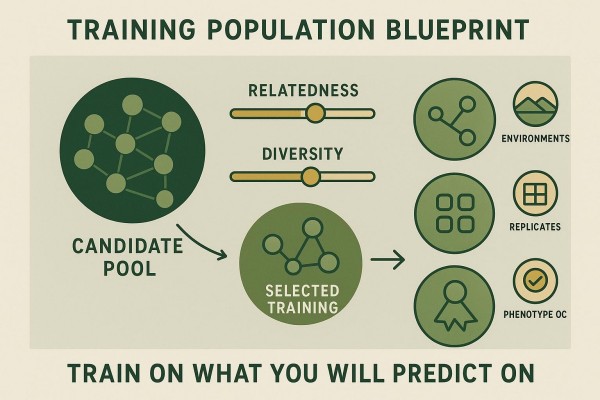
Training Population Design Drives Most of the GS Value
Prediction accuracy is controlled more by training population quality and phenotyping strategy than by model choice alone. Said differently, MAS and genomic selection will only deliver together if the GS half is trained on the right data.
Training Set Composition (Relatedness, Diversity, and Use-Case Alignment)
High realized accuracy depends on a training set that represents deployment germplasm and environments. Diverse but related training panels capture the haplotypes that candidates carry. Reviews across fruit and vegetable crops emphasize that right-sizing the training population and tuning relatedness to the prediction cohort matter more than squeezing out marginal gains from complex models—see Lee et al. (2024). The practical principle is simple: train on what the program will predict on.
Phenotype Strategy (Multi-Environment, Repeatability, Trait Definition)
Multi-environment trials (METs) with well-defined trait protocols improve both model fit and generalization. Studies in wheat and sesame show that multi-environment or multi-trait multi-environment models can raise predictive ability relative to single-environment fits when cross-environment correlations are positive (Gill et al., 2021; Sabag et al., 2023).
Confounding Control (Structure, Family Effects, and Environment)
Confounding erodes credibility. Programs should control family effects, include environment covariates where appropriate, and always visualize population structure (e.g., PCA) colored by batch and cohort to detect unintended shifts before model training.
Marker Strategy in a Hybrid Program
Hybrid success depends on separating "decision markers" (for MAS) from "genome-wide markers" (for GS) so each is optimized for its job.
Decision Markers: Robust Across Backgrounds and Batches
Decision markers must be robust across genetic backgrounds and lab batches. Functional markers or assays tightly linked to the causal variant are preferred; performance should be documented with call rate and concordance metrics and stress-tested against known genotype controls. For an accessible refresher on marker categories and their trade-offs, consult this overview of SSR, SNP, and GBS markers in MAS. Cross-crop generality matters too; for example, disease resistance MAS examples in tomato illustrate portable principles around validation and deployment, as outlined in this tomato disease-resistance MAS resource.
Genome-Wide Markers: Dense Enough for Prediction, Cheap Enough for Scale
For GS, density and stability trump perfection. At deployment scale, LC-WGS with imputation often yields high-density genotypes at a manageable cost when a representative reference panel exists. Evidence from animal and aquaculture genetics indicates that low-coverage sequencing (e.g., approximately half-fold coverage) combined with imputation can reach high concordance to high-density genotypes when references are well matched, as shown in rainbow trout evaluations that reported imputation r² above 0.95 in favorable settings (Liu et al., 2024). Conceptual primers in livestock underscore the same prerequisites—strong references, related cohorts, and careful QC (Rowan, 2025).
When to Convert Discovery Signals Into Routine Assays
Convert a discovery QTL into a routine MAS assay only after replicated effects across environments, cross-background assay robustness, and absence of deleterious linkage in the target germplasm are documented. Hybrid pipelines benefit most when very few, very reliable decision markers are promoted into the gate.
Genotyping Platform Choices for Hybrid Workflows
Platform selection should match stage: discovery and reference building differ from routine candidate screening at scale. A balanced, stage-aware view helps teams avoid chasing trends.
Discovery and Reference Panels (WGS and High-Density Genotyping)
Early work favors whole-genome or high-density methods to discover variants and establish reference panels that capture program diversity. Long-read scaffolding, where available, improves variant resolution around complex loci.
Routine Screening (SNP Arrays/Panels, GBS, or LC-WGS With Imputation)
For deployment, many programs now emphasize LC-WGS with imputation to drive GS, plus a small set of MAS decision assays. Arrays offer excellent cross-batch reproducibility for stable, long-lived pipelines, while GBS can be economical early on but may show higher missingness and enzyme-related batch effects. A neutral comparison of these options is summarized here: LC-WGS vs GBS vs SNP arrays for genomic selection. When a program wants practical details on LC-WGS implementation, platform and workflow considerations are outlined on this LC-WGS service page for further reading.
Practical Trade-Offs (Cost, Turnaround, Missingness, Comparability)
Public, program-scale head-to-head studies remain limited post-2020, but certain patterns are consistent. LC-WGS with imputation provides dense, flexible genotypes if a well-curated, related reference panel exists; arrays excel in reproducibility but are less flexible for new variants; GBS is adaptable and cost-effective early yet can challenge cross-batch comparability. Imputation accuracy depends on reference size/diversity, relatedness, LD extent, and MAF distributions; crop studies suggest performance gains plateau once certain density and reference composition thresholds are reached (Chen et al., 2022 and the broader imputation strategy guidance of Jiang et al., 2022).

Operational Governance: Prevent Model Drift and Marker Drift
Hybrid pipelines stay reliable only when marker sets, reference panels, and models are versioned, batch effects are monitored, and retraining occurs on a defined cadence—or when triggers fire.
Versioning Rules (Marker Sets, Reference Panels, Models, and Reports)
Use semantic versioning for decision marker panels and GS cores; publish changelogs with impact notes on cross-batch comparability. Freeze versions during deployment windows to avoid mid-cycle drift. Apply the same rigor to reference panels and model releases, keeping report templates synced to the active versions.
Monitoring Signals (Prediction Reliability, Batch Shifts, Trait Definition Drift)
Watch batch metrics (call rate, per-locus missingness, MAF and Hardy–Weinberg checks) and visualize PCA by batch to detect shifts. Track realized predictive ability on rolling validation cohorts and investigate drops against environmental changes or trait-definition adjustments. Concerns around GS reproducibility and the stability of complex models have been discussed conceptually in recent work, favoring transparent, auditable pipelines (see Howard et al., 2025).
Re-Training Triggers (New Germplasm, New Environments, New Phenotyping Protocols)
Common retraining triggers include the introduction of new germplasm that falls outside the current reference diversity band, new target environments or phenotyping protocols, and a sustained decay in realized predictive ability below the program's acceptance band.
Practical Deliverables and Acceptance Criteria
Decision-ready outputs should combine MAS calls, GS rankings, QC evidence, and a clear "why this was selected" audit trail—so that both breeders and program managers can act confidently.
Deliverables Checklist (Decision Markers, GEBVs, QC Summary, Selection List)
A standard release typically bundles: (1) MAS results for decision loci with assay IDs and genotype calls; (2) GS ranks with GEBVs and uncertainty bands; (3) QC summaries covering sample identity, batch comparability, and imputation quality; and (4) a selection list with rationale.
Acceptance Criteria (What Must Be True Before Acting on Results)
The following ranges are anonymized operational examples, not universal standards. Decision markers should achieve complete call rates and high cross-batch concordance; genome-wide data should meet per-sample call-rate and post-imputation missingness bands consistent with prior batches; shared-core marker overlap should exceed the program's comparability threshold; PCA should reveal no unexplained batch separation; and the current model release should meet or exceed the program's minimum realized predictive ability on a rolling validation cohort.
Communication Templates for PMs and Breeders (One-Page Brief)
One-page briefs work best when they place the MAS gate results and GS ranks side by side, summarize QC status with a compact dashboard, and state the advancement rationale in plain language.
How CD Genomics Can Support Hybrid MAS + GS Programs
A specialized CRO can provide genotyping and project-ready outputs that align with the hybrid workflows described above. For example, CD Genomics supports discovery and reference-panel development, routine LC-WGS with imputation for polygenic prediction, and compact MAS assay panels for validated decision loci, delivered for research use only (RUO). The practical value lies in harmonizing lab metrics with modeling gates—so that batch comparability, imputation quality, and version alignment are visible in the same deliverable. Teams exploring service options can review the broader offering on the agricultural services hub; inquiries typically include trait context, population scope, scale, timelines, and success criteria so the workflow can be shaped to existing QC gates and reporting requirements . Where desired, gates and report templates are aligned before the first cohort to prevent mid-project drift.
FAQ
When should MAS, GS, or a hybrid strategy be used for a complex trait?
- A program should use MAS when a small number of validated, large-effect loci control material risk or benefit (for example, rice blast R genes), and use GS when many small effects drive outcomes such as yield stability. A hybrid approach is recommended when both conditions hold, with MAS acting as a safety gate and GS providing early, genome-wide ranking across environments, a pattern consistent with recent reviews of integrated approaches and training-population priorities.
How can a team decide whether a locus is "validated enough" to use for MAS inside a GS pipeline?
- The bar is crossed when assays are robust across genetic backgrounds and lab batches, phenotypic effects replicate across sites and years or on an independent validation panel, effect sizes are deployable in target backgrounds, and no deleterious linkage is evident. Operationally, this means documenting call-rate and concordance metrics and demonstrating stability in representative germplasm before promoting a locus into a decision gate.
What training population size and design choices most improve GS prediction accuracy?
- Realized accuracy responds most to using a training set that aligns with the candidates and the target environments, to increasing the training set to capture relevant haplotypes, and to investing in multi-environment phenotyping with repeatability. Model selection matters, but less than these data choices; multi-environment or multi-trait models often add value when cross-environment correlations are positive.
Which genotyping platform is most cost-effective for hybrid programs at scale (LC-WGS, GBS, SNP arrays)?
- Many programs select LC-WGS with imputation for deployment because it delivers dense, flexible genotypes when a solid reference panel is available; arrays remain attractive for pipelines that prize cross-batch reproducibility and long-term stability; GBS can be cost-effective early on but may complicate cross-batch comparability due to enzyme and library effects. The best choice depends on reference maturity, comparability requirements, and budget at scale.
How often should genomic prediction models be retrained and marker sets updated to avoid drift?
- Teams retrain on a cadence tied to breeding cycles or when triggers fire—introduction of new germplasm, new environments or phenotyping protocols, or a sustained drop in realized predictive ability below a program-defined threshold. Version all changes, publish concise release notes, and freeze versions during active deployment windows to preserve comparability.
References
- Feng, Zhiming, et al. "Development of Rice Variety With Durable and Broad-Spectrum Resistance to Blast Disease Through Marker-Assisted Introduction of Pigm Gene." Frontiers in Plant Science, 2022.
- Thulasinathan, Thiyagarajan, et al. "Marker Assisted Introgression of Resistance Genes and Phenotypic Evaluation Enabled Identification of Durable and Broad-Spectrum Blast Resistance in Elite Rice Cultivar, CO 51." Genes, 2023.
- Asekova, S., et al. "Multi-environment analysis enhances genomic prediction accuracy of agronomic traits in sesame." Frontiers in Genetics, 2023.
- Gill, Harsimardeep S., et al. "Multi-Trait Multi-Environment Genomic Prediction of Agronomic Traits in Advanced Breeding Lines of Winter Wheat." Frontiers in Plant Science, 2021.
- Jiang, Y., et al. "Exploring the optimal strategy of imputation from SNP array to whole-genome sequencing data in farm animals." Frontiers in Genetics, 2022.
- Pavan, S., et al. "Recommendations for Choosing the Genotyping Method and Best Practices for Quality Control in Crop Genome-Wide Association Studies." Frontiers in Genetics, 2020.
- Liu, Sixin, et al. "Accurate genotype imputation from low-coverage whole-genome sequencing data of rainbow trout." G3: Genes|Genomes|Genetics, 2024.
- Heaton, Matthew P. "A primer on sequencing and genotype imputation in cattle." Journal of Animal Science, 2025.
 Send a Message
Send a MessageFor any general inquiries, please fill out the form below.


