
Marker-Assisted Selection in Animal Breeding: Study Design, Confounding Control, and Use Cases

Modern livestock programs are under pressure to make faster, cleaner decisions with fewer errors. For traits influenced by known major loci and for identity or pedigree verification tasks, marker-assisted selection offers a targeted path—provided the study design resists confounding, and the data pass auditable QC gates. This guide explains how to apply marker-assisted selection in animal breeding across cattle, pigs, poultry, and small ruminants, with a practical emphasis on study design, confounding control, cross-breed validation, and decision-ready deliverables.
Key takeaways
- Marker-assisted selection in animal breeding is most effective for validated major-effect loci and identity workflows such as parentage and breed composition, while genomic selection remains preferred for highly polygenic traits.
- Study validity relies more on design choices and confounder control than on any single lab platform; freeze acceptance criteria and QC gates before genotyping.
- Cross-breed validation is essential because LD phase and allele frequencies differ across breeds and crossbreds; a validation ladder protects against overfitting.
- Decision-ready outputs combine genotype and annotation with transparent QC evidence, applicability bounds, and a clear table of next-step actions.
What MAS means in animals
One-sentence definition Marker-assisted selection in animals uses a small number of validated genetic markers linked to major-effect loci to enable earlier, more accurate selection decisions while controlling for family structure and population stratification.
For foundational concepts and workflows, see the primer on marker-assisted selection in the context of molecular breeding in the article on the MAS overview from CD Genomics: marker-assisted selection MAS.
MAS vs genomic selection as a boundary
MAS focuses on a compact set of validated markers tied to large or decisive effects. Genomic selection models genome-wide SNPs to capture the additive variance of complex traits. For polygenic traits, genome-wide prediction generally wins on accuracy; for identity tasks and major loci, MAS can provide fast, interpretable decisions. Readers seeking deeper coverage of genomic selection can consult the internal resource outlining concepts, training populations, and modeling approaches here: genomic selection in plant and animal breeding.
Practical limits and safe claims in research settings
Evidence standards matter. Claims should reflect replicated effects with clear applicability bounds. When validating or deploying markers, publish the intended population scope, allele frequencies, and assumptions. Where uncertainty remains, favor a hybrid approach that treats major loci using targeted markers while retaining genome-wide information for polygenic background.
Decide before genotyping
Decision timing, trait architecture, and error cost determine whether marker-assisted selection is appropriate more than any single lab method. A brief comparison of molecular and non-molecular pathways is available in the internal primer contrasting strategies: MAS compared with phenotypic selection.
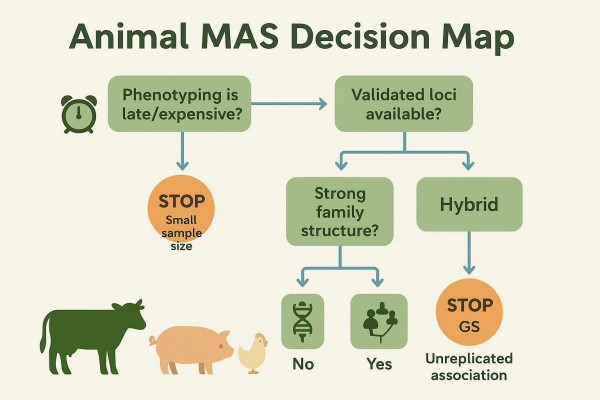
Trait architecture major locus versus polygenic background
- Major locus traits or identity tasks fit MAS when a limited number of markers consistently track the biology of interest. Think quality-assured parentage, breed composition bounds, or deployment of a well-validated major QTL.
- Polygenic traits such as yield, growth, and fertility respond better to genome-wide models. In these cases, marker-assisted selection should not serve as the primary decision system.
When MAS fits and when it misleads
MAS fits when phenotyping is late or expensive, when strong and replicated loci are available, and when selection errors would be costly. It misleads when the evidence base is thin, when alleles are near-fixed or rare within the deployment population, or when family structure causes apparent signals that are not causal. Conservative "stop" conditions include weak replication, ambiguous LD phase across breeds, and untested cross-batch comparability.
Evidence required for selection-ready markers
Markers should pass discovery QC, replicate in additional breeds or representative crossbreds, and demonstrate stable effect direction and magnitude under varied management contexts. Report allele frequencies and LD context, and pre-register the decision rule that links genotype to action.
Study designs that work
Animal MAS designs must handle relatedness and stratification so that markers track biology rather than family lines or management artifacts. A concise overview of breeding pipeline components is available here: genomic breeding services overview. For population structures frequently used in research and breeding, see the guide to common populations: genetic breeding populations resource.
Family-based designs in structured herds
In half-sib or full-sib designs and structured herds, within-family contrasts reduce confounding. Designs that block on sire or dam, combined with pedigree-informed models, ensure that observed associations are not due merely to shared ancestry. Trio or duo information enables Mendelian checks that quickly surface swaps and contamination.
Population designs across breeds and crossbreds
Multi-breed and crossbred cohorts require tolerance for heterogeneous LD and background effects. Kinship-aware association models and balanced sampling by breed prevent confounding between ancestry and marker signal. When possible, designs should ensure connectivity among herds and lines to support external validation.
Decision unit individual versus family versus line
Define the decision unit before sampling begins. Identity and pedigree QC decisions typically occur at the individual level. Introgression or backcross strategies may decide at the family or line level. The definition of decision unit determines sample sizes, blocking, and the reporting format of the final decision table.
Control confounding
Confounding control prevents false signals caused by ancestry, relatedness, or management rather than true trait biology. Many livestock GWAS design and QC practices are directly useful for MAS discovery and validation, especially for controlling structure, relatedness, and batch effects. The 2024 invited review on quality control in animal breeding programs outlines common QC expectations and diagnostics relevant to both GWAS and MAS according to the Journal of Animal Science and Technology in the article titled quality control strategies and applications in animal breeding programs (2024).
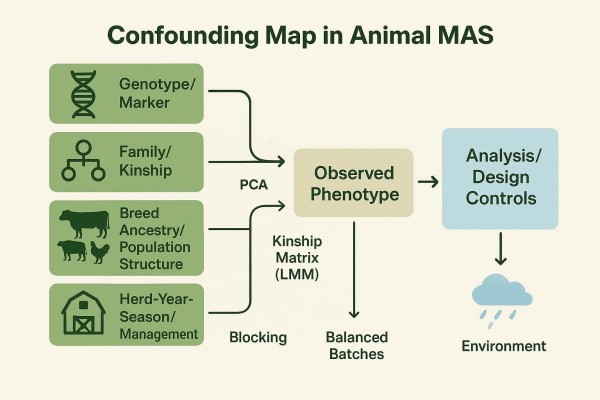
Population structure and breed stratification
Principal component analysis is a practical first step to profile structure and to visualize breed or line clusters. Including appropriate PCs in association models helps reduce inflation from stratification. In crossbred contexts, breed intercepts and breed-specific allele effects may be required to stabilize inference. Methodological blueprints from crossbred genomic evaluation research describe how unified SNP effect sets and breed adjustments are estimated across mixed populations in the open-access review on across-breed genomic prediction and validation published in 2022.
Relatedness and kinship effects
Linear mixed models with a genomic relationship matrix absorb background relatedness and help separate marker signal from family effects. Kinship-aware modeling is particularly important in structured herds with strong half-sib clusters, where naïve models tend to overstate significance.
Herd year season and management factors
Management and environmental covariates such as herd, year, season, diet, and housing influence phenotypes. Blocking by herd-year-season and including contemporary group effects reduces confounding between management and genotype.
Do-not-skip controls in study setup
Balanced batches, blinded sample handling, independent technical replicates, and fixed control samples across runs prevent plate and operator effects from creeping into analysis. Prospective fold definitions that isolate by herd or line avoid leakage between training and testing sets.
Validate markers across breeds
Selection-ready markers require cross-breed validation because LD phase and allele frequencies change across populations.
Evidence sources for marker discovery
Linkage and QTL studies prioritize family-based signals, while association studies evaluate population-level correlations. For concept refreshers and method references, see the internal explainer on association mapping for trait dissection and the outline of gene mapping workflows in animals and plants. Foundational linkage mapping concepts are summarized in the primer on genetic linkage map construction.
Cross-breed robustness checks effect LD and frequency
Three minimum checks enable robust transfer:
- Effect direction and magnitude are consistent across the target breeds or representative crossbreds.
- LD phase between the marker and the causal variant is stable across populations. A common approach correlates signed square roots of r² across breed pairs along distance bins, as described by Frontiers in Genetics in a methodological overview on cross-population LD phase consistency (2021).
- Allele frequencies support decision utility in the deployment populations; near-fixed or ultra-rare alleles provide little value.
A validation ladder from discovery to deployment
- Discovery under strict QC and confounder control.
- Replication in additional pure breeds and representative crossbreds.
- Robustness checks under varied management and climate, including LD phase and frequency surveys.
- Prospective pilot decisions with tracked outcomes.
- Deployment in a locked assay with documented performance bounds and applicability notes.
Choose markers and platforms
Platform choice should match the goal: reproducible decision markers for deployment versus broad discovery data for marker finding. Operational comparisons are summarized in the internal guide on SSR versus SNP versus GBS for MAS.
SSR and SNP and sequencing in practice
- SSRs serve legacy roles in some registries and are still used in parentage where SNP infrastructure is limited, but issues such as null alleles and stutter are well documented.
- SNP panels have become the default for routine parentage and many MAS deployments due to stability, scalability, and clean cross-batch comparability.
- Reduced-representation sequencing or low-pass WGS coupled with imputation is valuable for discovery and for species or breeds lacking mature arrays.
Throughput and cross-batch comparability
For deployment, favor fixed panels with locked assay conditions and control samples that travel across batches. Define harmonization procedures and track cross-run concordance explicitly.
Discovery versus deployment data strategies
Discovery benefits from broader coverage, such as whole genome resequencing or low-pass WGS, to fine-map and to build reference panels. For routine screening and decisions, lock a small, validated panel with explicit QC gates. For discovery modalities and their roles in building training sets, see the internal explainer on animal and plant whole genome sequencing.
QC gates for decision-ready data
Decision-ready MAS data require identity checks, reproducible calls, and batch comparability before generating any selection list. The 2024 review in the Journal of Animal Science and Technology details common thresholds and diagnostics for livestock genotyping pipelines in the article on quality control strategies and applications in animal breeding programs (2024). Low-coverage sequencing with imputation has been benchmarked across populations, with accuracy gains when reference panels are breed-matched as summarized by PLoS Computational Biology in the open-access paper on low coverage sequencing and imputation performance (2023).
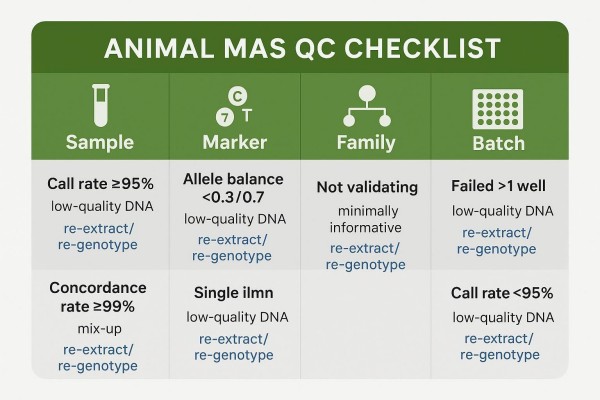
Below is a compact table of reference bands. These reference bands should be tuned to platform behavior, species genetics, batch design, and the downstream decision risk defined in your protocol..
| QC gate | Metric or rule | Typical reference band | Why it matters | Action when failing |
|---|---|---|---|---|
| Sample | Call rate | ≥ 90–95% | Low-quality DNA or assay failure | Re-extract or re-genotype; remove if persistent |
| Sample | Identity and duplicates | 100% concordance across replicates | Detect swaps and contamination | Resolve identity; repeat; document exclusions |
| Marker | SNP call rate | ≥ 95–99% | Unreliable loci inflate noise | Exclude low-performing SNPs |
| Marker | Minor allele frequency | ≥ 0.01–0.05 context dependent | Extremely rare alleles add variance without utility | Filter or note reduced utility |
| Marker | Hardy–Weinberg checks | Remove extreme deviations after structure accounted | Detect genotyping artifacts | Review clustering; drop problematic loci |
| Family | Mendelian errors | Near-zero for validated trios | Detect swaps and pedigree errors | Investigate, correct pedigree, or exclude |
| Batch | Plate or run effects | No cluster separation by batch in PCA | Prevent confounded calls | Harmonize; include traveling controls; reprocess |
A chicken-focused example using a 0.05 MAF threshold illustrates typical population filters in poultry genetics according to Poultry Science in the study on genomic analysis and filtering in chicken cohorts (2024).
Where animal MAS adds value
Marker-assisted selection creates the most value when decisions are frequent, error cost is high, and markers are validated for deployment.
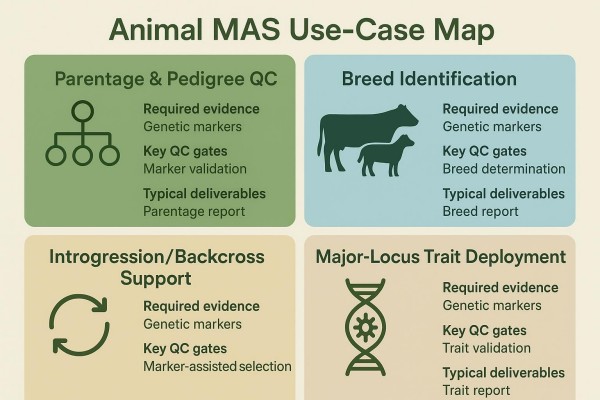
Parentage and pedigree QC
Parentage and pedigree verification is a high-frequency, cross-species workflow with mature standards in cattle and growing resources in other species. Programs typically publish exclusion probabilities for duo and trio contexts, mismatch handling rules, and identity controls. ICAR's certification framework, aligned with ISAG comparison tests, outlines expectations for cattle, including declared marker sets and performance disclosure as summarized on the ICAR site in the page describing the 2025 certification for genetic laboratories by SNP and STR methods (2025). For pigs, small ruminants, and poultry, panel composition and acceptance criteria remain registry- and country-specific; current public, post‑2020 core SNP standards are less consolidated, so programs should verify species working group updates before locking deployments.
Breed identification and composition
Breed identification and composition are common identity tasks that rely on breed-informative marker sets and supervised classifiers with breed-aware cross-validation. An overview of marker discovery and assignment workflows is provided here: breed identification and composition.
Introgression and backcross support
When a favorable allele is introduced from a donor line, MAS can track the target allele while background selection steers the genome back toward the recurrent line. Decision units may be families or lines, with checkpoints that verify both target-locus status and genome-wide recovery.
Deploying major loci for applied traits
For well-validated major QTL with consistent effects across deployment populations, MAS provides fast, interpretable selection that is easy to communicate to non-technical stakeholders. Applicability notes should include validation breeds, expected effect ranges, and any known gene-by-environment interactions.
Deliverables that teams use
Actionable deliverables translate genotypes into a selection-ready table backed by QC evidence and clear next steps.
What to deliver in a decision package
A standard package includes genotype calls for the decision markers, succinct variant annotation and biological rationale, cross-breed validation notes, and a decision table that maps genotype state to action. For programs running broader molecular breeding or genotyping projects, a summary of upstream methods and data structures is helpful; a concise overview is available here: molecular breeding and genotyping workflows.
Metadata that prevents rework
Pedigree, herd-year-season, trait definitions, and management context allow downstream analysts to interpret results accurately and to reproduce decisions later. Identity keys and stable sample IDs prevent "lost-in-translation" errors between lab, bioinformatics, and breeding teams.
Acceptance checklist for internal sign-off
The acceptance checklist should link each QC gate to a go or no-go outcome, with corrective actions and documentation requirements. Final archives should include raw and processed data, parameter files, software versions, and a changelog.
How CD Genomics can help
As an end-to-end agricultural genomics provider, CD Genomics can support discovery-to-deployment pipelines for marker-assisted selection and identity workflows across species. Typical collaborations include designing discovery studies, validating markers across breeds, and producing decision-ready panels and reports for research use only. When projects involve ongoing routine screening, teams can align on QC gates, traveling controls, and reporting formats up front. A summary of cross-platform capabilities is available here: agricultural genomics services overview. Any laboratory support described in this section is intended for RUO contexts.
FAQ
Q1: When should MAS be used instead of genomic selection in animal breeding?
A: MAS is typically most useful when you have validated loci with actionable effect sizes and you need a clear, auditable decision gate (pass/fail/retest). Genomic selection is often preferred for complex traits driven by many small effects, where ranking candidates early provides more value than fixing a single locus.
Q2: How do I control for family relatedness and population structure in animal MAS studies?
A: Start with design controls (balanced sampling across families/herds/breeds, clear decision unit, and consistent phenotyping), then confirm with analysis checks (PCA/structure diagnostics, kinship-aware models, and sensitivity analyses). Treat "marker tracks a family line" as a common failure mode and require cross-family validation before deployment.
Q3: What evidence is needed to deploy a marker across breeds or crossbreds?
A: A deployment-ready marker should show consistent direction of effect across backgrounds, acceptable allele frequency for decision-making, and robustness under the environments and management systems that match your target population. A practical ladder is: discovery → replication → cross-breed validation → multi-batch reproducibility → operational deployment.
Q4: Which QC checks should be mandatory before using genotypes for selection decisions?
A: At minimum, enforce sample identity QC (duplicates/swaps/outliers), marker QC (missingness and concordance), family QC where pedigrees exist (Mendelian consistency), and batch QC (plate/run effects and cross-batch comparability). Pair each QC metric with a defined action: accept, retest, re-extract, or exclude.
Q5: What deliverables make MAS results decision-ready for breeding teams?
A: Decision-ready deliverables include (1) a genotype dataset in standard formats, (2) marker annotation and interpretation notes, (3) a QC summary with pass/fail flags, and (4) a genotype-to-action decision table aligned with your breeding rule set. Versioning notes for marker sets and batches help keep decisions comparable over time.
References
- Lee, Jungjae, Jong Hyun Jung, and Sang-Hyon Oh. "Enhancing Animal Breeding through Quality Control in Genomic Data—A Review." Journal of Animal Science and Technology, 2024.
- Misztal, I., Y. Steyn, and D. A. L. Lourenco. "Genomic Evaluation with Multibreed and Crossbred Data." JDS Communications, vol. 3, no. 2, 2022, pp. 156–159.
- Pasaniuc, Bogdan, et al. "Efficient Methods for Combining Sequence and SNP Genotype Data in Association Studies." PLOS Computational Biology, 2012.
- Gebrehiwot, Sewalem, et al. "Inference of Ancestries and Heterozygosity Proportion and Genotype Imputation in West African Cattle Populations." Frontiers in Genetics, vol. 12, 2021, Article 584355.
- Yan Zhou, et al. "Pedigree Reconstruction Based on Genotype Data in Chickens." Poultry Science, 2024.
 Send a Message
Send a MessageFor any general inquiries, please fill out the form below.


