GWAS in Agriculture: Application Cases and Impacts on Agricultural Development
GWAS in Agriculture: Application Cases and Impacts on Agricultural Development
Inquiry
With the vigorous development of agriculture and the constant pursuit of high yield and high quality, genetic research is the key driving force to break through the bottleneck of agricultural production. Genome-wide association study (GWAS) technology is just like a precise key, which opens the door to deeply understand the genetic mystery of crops and livestock. From analyzing the high-yield genes of wheat to exploring the genetic markers related to high-quality milk production of dairy cows, GWAS has produced many achievements in the agricultural field. Through specific learning cases, we can understand how to innovate agricultural scientific research and production practice.
Deeply Analyze the Evolutionary Mechanism
In modern agricultural science, it is very important to deeply analyze the genetic basis and evolutionary mechanism of crops to improve agricultural productivity and food security. The study of population evolution combined with GWAS provides us with a powerful method to reveal the genetic variation and adaptability of crops under natural selection and artificial selection. The study of population evolution can track the historical dynamics of different varieties and populations and analyze their evolutionary trajectories under different environments and selection pressures. By analyzing large-scale genome data, GWAS can accurately locate the genetic variation affecting important agronomic traits and find key gene loci related to disease resistance, yield and quality. This comprehensive method not only improves our understanding of crop genetic diversity and evolutionary mechanism, but also provides scientific basis for precision breeding, promotes the cultivation of new crop varieties with high yield, stress resistance and high quality, and then promotes the sustainable development of agriculture and the realization of global food security.
Carrot (Daucus Carota L.) is famous for its rich dietary vitamin A-protocarotenoids, α-carotene and β-carotene, and its output has steadily increased in the past 50 years. Carrot germplasm resource bank includes various cultivated varieties, local varieties and wild carrots, which has extensive phenotypic diversity and can be used for breeding. Carrots are mainly divided into east and west. The east carrot is the earliest domesticated purple or yellow carrot, which originated in Asia Minor and Central Asia. Western carrots are mainly orange, which first appeared in Europe in the 17th century, and quickly became the main carrot type planted and consumed in the world. In order to better understand the process of carrot domestication and improvement and its influence on gene diversity, it is very important to study population evolution and GWAS.
By resequencing 630 carrot germplasm resources, 25,375,112 SNPs were identified. Population structure analysis identified five main populations, namely wild population, local variety A, local variety B, early cultivated variety and improved cultivated variety. Genetic diversity analysis showed that the nucleotide diversity of wild carrots was the highest, while the cultivated varieties were the lowest. The decay rate of LD in cultivated carrot population was slow, which indicated that the genetic diversity decreased significantly after domestication and improvement. The historical results of population size support the genetic bottleneck and recent expansion in the process of domestication and improvement. Through GWAS analysis, the significant association sites related to carotenoid accumulation on chromosomes 2, 3 and 7 were identified, and key genes including DCAR_730022 and DCAR_310369 were identified. This study provided important genetic resources and theoretical support for carrot breeding.
Genetic diversity and demographic analysis of carrot germplasm (Coe et al., 2023)
Maize is one of the important food crops in the world, and its hybrid breeding is of great significance in improving yield and adaptability. However, the molecular mechanism of heterosis is not completely clear, especially in complex hybrid populations, the role of gene regulatory networks and key genes still needs to be further studied.
A hybrid population was designed and constructed by North Carolina II (NCII), and 78 female parent lines were crossed with 4 male parent lines, resulting in 314 hybrid lines. By re-sequencing the whole genome of 82 parental lines, 4,628,240 high-quality SNPs were identified. Phenotypic analysis showed that there was a strong correlation between plant height (PH) and ear height (EH), which indicated that the phenotypic data was reliable. The comparison between hybrid performance and mid-parent phenotype showed that all hybrid lines showed strong heterosis for these two traits. The generalized heritability of plant height and ear height is 0.96, while the heterosis of middle parents is 0.70 of plant height and 0.77 of ear height, respectively, indicating that the hybrid lines are stable in different environments.
GWAS analysis identified 11 additive-QTLs and 41 dominant-QTLs related to plant height, while 21 additive-QTLs and 6 dominant-QTLs related to ear height. Comparing the physical positions of QTLs for plant height and ear height, it was found that one additive -QTL and three dominant-QTLs were related to both traits. The phenotypic variance interpretation rate (PVE) of dominant -QTL was significantly higher than that of additive -QTL. Further screening annotation genes in QTL region or reported homologous genes related to plant growth or development as candidate genes. Six additive-QTLs and 15 dominant-QTLs related to plant height, and eight additive-QTLs and four dominant-QTLs related to ear height contain candidate genes related to plant growth or development. The cumulative effect of favorable QTL genotypes (called "favorable scores") and hybridization performance have significant correlation with the two traits, which supports the reliability of these QTLs. In addition, the heterozygosity genotype of dominant QTL significantly increased the heterosis of parents, while additive QTL showed allelic dose effect.
Phenotypic and genetic analysis of plant height and ear height in hybrid population (Zhang et al., 2025)
Bring New Thoughts in Precision Agriculture
Genetic and environmental factors jointly determine the growth and yield of plants. In the past 20 years, GWAS have been conducted on crops to decipher the genetic loci that promote growth and yield. However, plant genotype seems to be insufficient to explain the trait variation. Using the methods of comprehensive GWAS, microbial-wide association studies (MWAS) and microbial-genome-wide association studies (MGWAS), the researchers revealed the correlation between genotypes, phenotypes and root surface microbial variables of 827 millet varieties.
A total of 827 millet varieties were collected for sequencing and SNP information was obtained. After strict quality control steps, including population stratification and pedigree filtering, individual and locus level filtering and minor allele frequency (MAF) filtering, a total of 161,562 SNPs were detected. SNPs were distributed evenly along chromosomes, and the genetic distance of linkage disequilibrium (LD) attenuated to half its maximum value was 9 kb. Phylogenetic analysis based on genetic SNPs showed that there were three main groups of millet varieties tested.
Genotypic phenotypic analysis showed that all the other 11 traits had significant heritability (H2=0.006, P=0.15), and the heritability of growth traits was higher than that of yield traits, among which MSPD had the highest heritability (H2 = 0.46, generalized heritability) and PGW had the lowest heritability (H2 = 0.16). The phenotype was GWAS to determine the SNP related to growth and yield traits. Under the threshold of P value, 86 significant SNP loci and 91 association results of 10 traits (except MSPW and TSLW) were identified, among which some SNP loci were related to multiple traits, among which 15,16,11,10 and 16 significant SNPs were located on chromosomes 2,4,6,7 and 9, respectively.
GWAS of agronomic traits in foxtail millet (Wang et al., 2022)
Analyze Protein Grain Content in Rice
Grain Protein Content (GPC) is the key factor to determine the nutrition and eating quality of rice. Obtaining rice GPC quickly and accurately and identifying related genes is of great significance for analyzing its genetic basis and breeding high-quality rice varieties.
The researchers compared and analyzed the GWAS results of measured GPC, GPC estimation based on original data (PLSR model based on preferred wavelet features when the training data is 276) and GPC estimation based on simulation data (PLSR model based on preferred wavelet features when the simulation data is 200). From the figure, it can be seen that all three of them are located at the same site (SNP 12.56465), and the estimated value 2 coincides with the measured GPC and the estimated value 1 with two lead SNPs, namely SNP12.5076465, SNP7.10830236, SNP 12.465, SNP 5.5005108006 At the same time, it was found that only within ±100kb of SNP4.17571584 located by estimated value 2, the grain storage protein related gene OsmtSSB1L could be detected. By comparing the similarities and differences of the loci detected by the estimated value and the measured value, it can be concluded that the GPC predicted value based on DCGAN has a good ability to replace the measured value for genetic analysis.
The research shows that DCGAN can generate spectrum and simulated GPC data to solve the problem of limited measured data, and the simulated GPC distribution range is wider after 8000 iterations. Data enhancement can improve the accuracy of GPC estimation model, and the performance of PLSR model is the best when the simulation samples reach 200. GPC data estimated by DCGAN can replace the measured values for genetic analysis, and can detect the same loci as the measured values, as well as discover new loci and related genes.
Manhattan (A, C, and E) and quantile-quantile (B, D, and F) plots of measured GPC estimated value 1 and estimated value 2 (Zheng et al., 2024)
Develop New Resistant Variety
Rice (Oryza sativa L.) is one of the important food crops in the world. Pursuing high yield is the eternal breeding goal of rice breeding, but high-yield varieties often have poor quality and poor disease resistance, and the phenomenon of "high yield without disease resistance" or "high quality without high yield" widely exists in crop breeding process. Some japonica rice varieties in Central China are of poor quality or susceptible to rice blast, which seriously limits their application in rice production. Therefore, how to balance yield, quality and disease resistance and cultivate varieties with high yield, high quality and high disease resistance has become a problem for breeders.
In the paper, 200 japonica rice varieties planted in central China in recent 30 years were re-sequenced, and variation detection was carried out. It was found that the varieties from different regions had significant genetic diversity. In order to identify the key genes and intervals related to yield in modern rice breeding, the researchers identified four key agronomic traits in four environments, including yield per plant (YP), 1000-grain weight (GW), number of grains per plant (GN) and number of spikes per plant (PN), calculated the best linear unbiased prediction (BLUP) value, and analyzed the correlation among the four agronomic traits. Subsequently, GWAS analysis combined with resequencing data identified 20 loci related to these yield-related traits, of which 11 were previously reported and functional genes had been cloned, such as BG3, TGW6 and DEP1.
Combined with the previously identified AC phenotypes, the researchers divided all varieties into three types: rice blast sensitive and low amylose varieties (BSLVs), rice blast resistant and medium amylose varieties (BRMVs) and rice blast sensitive and medium amylose varieties (BSMVs). Three loci were identified by GWAS analysis, among which Piz was the most significant one. While most of the significant SNPs were distributed near the leucine-rich resistance gene (Os06g0286700). Finally, two key SNPs, Chr6:10389856 and Chr6:10389883, were identified. The genotypes of blast resistance alleles in Piz-t gene were "G" and "C", while the susceptible genotypes and Pi2, Pi9 and Pigm were "T" and "G" (Figure 3e). In addition, the author found that all 17 japonica rice varieties in Jiaxing, Zhejiang Province carried Piz-t allele except the intermediate material "Yangjing 7311", which indicated that Piz-t genotype was the main rice blast resistance gene of japonica rice varieties in Central China.
By analyzing the excellent allele frequency distribution of genes identified by GWAS analysis, it was found that compared with BRMVs and BSMVs, BSLVs aggregated more excellent alleles related to yield and excellent taste, but the main rice blast resistance gene Pizt/ Pigm was lost. The researchers speculated that this may be related to the genetic linkage between genes. Therefore, a genome-wide linkage map was constructed, and it was found that the distribution of linkage blocks in each chromosome was very uneven. Furthermore, the genetic linkage phenomenon between yield, taste and rice blast resistance genes on chromosome 6 was analyzed, and an important type of linkage resistance was observed. Although BRMVs, a highly resistant variety, carried the dominant allele of rice blast resistance, the dominant allele of quality and yield was missing. In the era of promoting high yield and high quality breeding, this will lead to the sharp disappearance of dominant major disease resistance alleles in breeding population.
In the study, firstly, based on the resequencing data of 200 japonica rice populations from Central China, GWAS analysis and XPCLR analysis were carried out, and excellent genes related to yield, eating quality and rice blast resistance were identified. Excellent taste quality and rice blast resistance alleles were successfully introduced into two high-yield varieties, and two excellent rice blast resistance lines XY99 and JXY1 with excellent taste, high yield and broad spectrum were cultivated.
GWAS of genes related to yield traits (Xiao et al., 2021)
Conclusion
The application of GWAS in agriculture has shown great potential. Many cases have achieved fruitful results, from accurately locating crop disease-resistant genes, helping to cultivate excellent varieties, to analyzing the genetic mechanism of important economic traits of livestock and improving breeding efficiency. Looking forward to the future, with the continuous innovation of technology, GWAS will be more deeply integrated into the whole agricultural industry chain. It will write a more brilliant chapter in responding to the global food security challenge, realizing the sustainable development of agriculture, ensuring the supply of agricultural products and optimizing the layout of agricultural production.
References
Coe, K., Bostan, H., Rolling, W., et al. "Population genomics identifies genetic signatures of carrot domestication and improvement and uncovers the origin of high-carotenoid orange carrots." Nat. Plants 9. 2023: 1643–1658 https://doi.org/10.1038/s41477-023-01526-6
Zhang J, Gu R., et al. "GWAS-based population genetic analysis identifies bZIP29 as a heterotic gene in maize." Plant Commun. 2025 20:101289 https://doi.org/10.1016/j.xplc.2025.101289
Wang Y, Wang X., et al. "GWAS, MWAS and mGWAS provide insights into precision agriculture based on genotype-dependent microbial effects in foxtail millet." Nat Commun. 2022 13(1):5913 https://doi.org/10.1038/s41467-022-33238-4
Zheng H, Tang W., et al. "Grain Protein Content Phenotyping in Rice via Hyperspectral Imaging Technology and a Genome-Wide Association Study." Plant Phenomics. 2024 6:0200 https://doi.org/10.34133/plantphenomics.0200
Xiao N, Pan C., et al. "Genomic insight into balancing high yield, good quality, and blast resistance of japonica rice." Genome Biol. 2021 22(1):283 https://doi.org/10.1186/s13059-021-02488-8
For research purposes only,
not intended for clinical diagnosis, treatment, or individual health assessments.
For any general inquiries, please fill out the form below.
We provide the best service according to your needs Contact Us
PDF Download
×
OUR MISSION
CD Genomics is propelling the future of agriculture by employing cutting-edge sequencing and genotyping technologies to predict and enhance multiple complex polygenic traits within breeding populations.

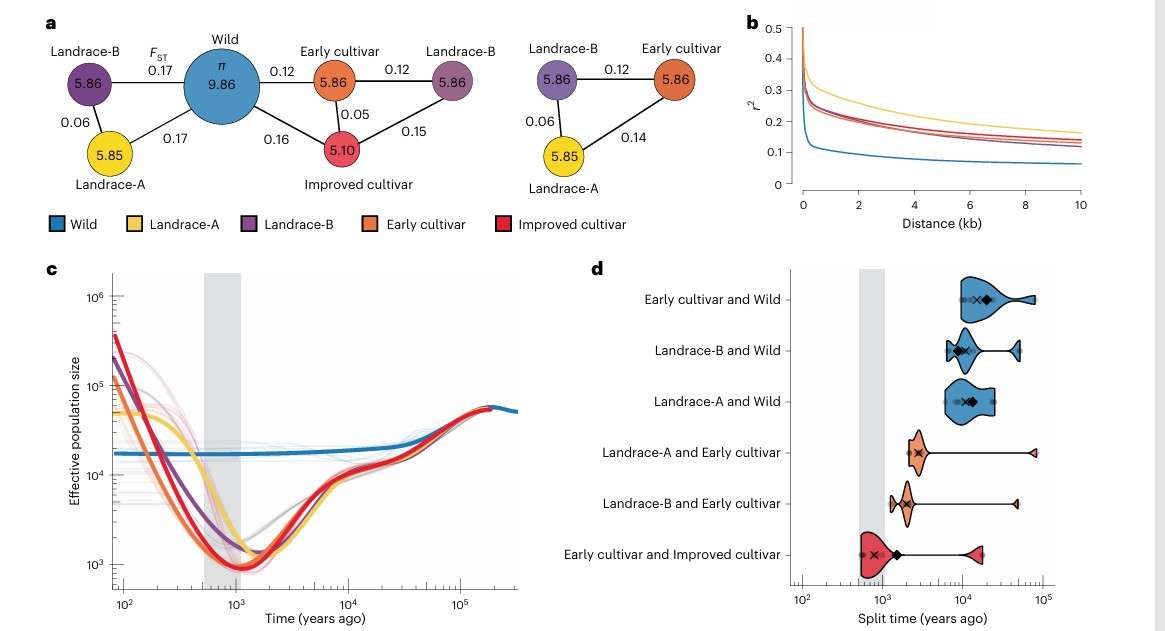 Genetic diversity and demographic analysis of carrot germplasm (Coe et al., 2023)
Genetic diversity and demographic analysis of carrot germplasm (Coe et al., 2023)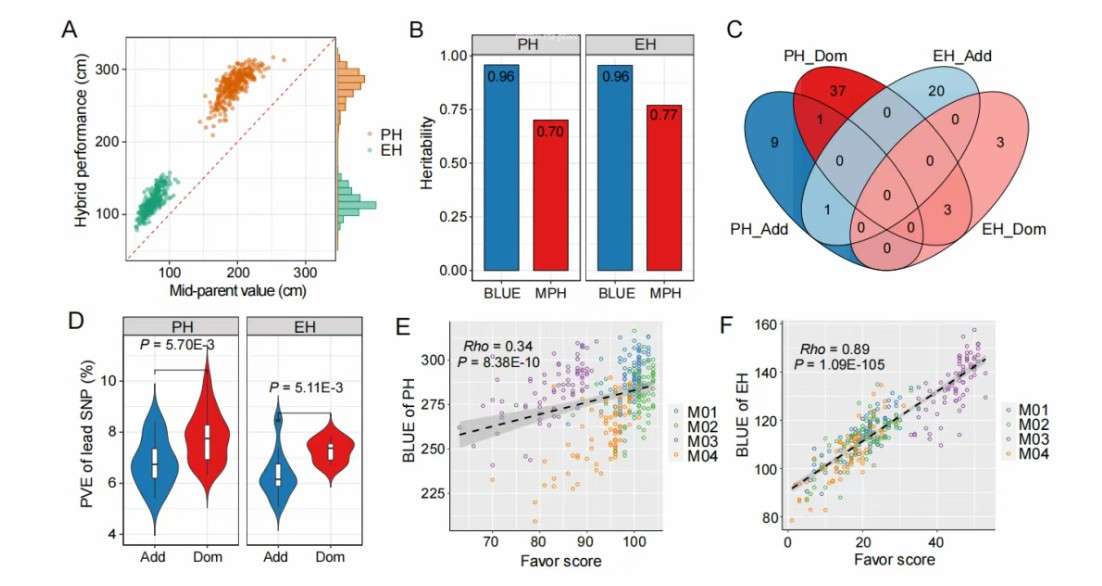 Phenotypic and genetic analysis of plant height and ear height in hybrid population (Zhang et al., 2025)
Phenotypic and genetic analysis of plant height and ear height in hybrid population (Zhang et al., 2025)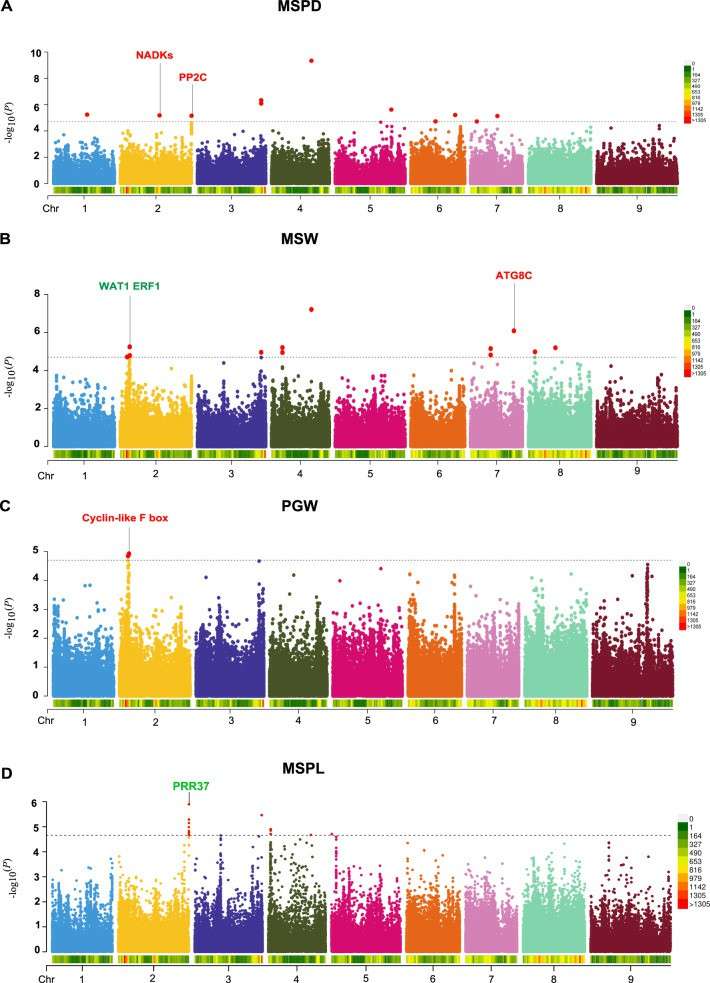 GWAS of agronomic traits in foxtail millet (Wang et al., 2022)
GWAS of agronomic traits in foxtail millet (Wang et al., 2022)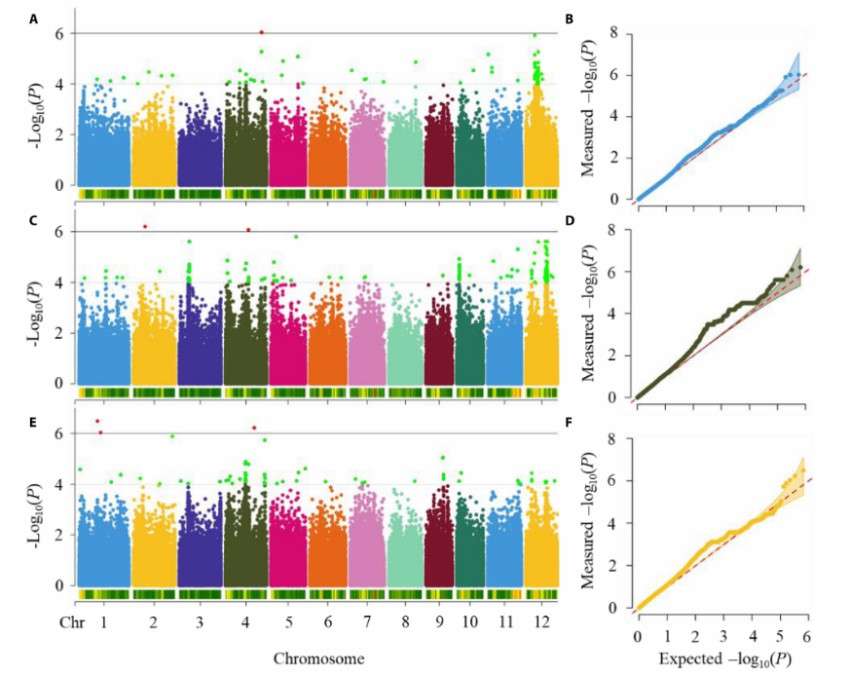 Manhattan (A, C, and E) and quantile-quantile (B, D, and F) plots of measured GPC estimated value 1 and estimated value 2 (Zheng et al., 2024)
Manhattan (A, C, and E) and quantile-quantile (B, D, and F) plots of measured GPC estimated value 1 and estimated value 2 (Zheng et al., 2024)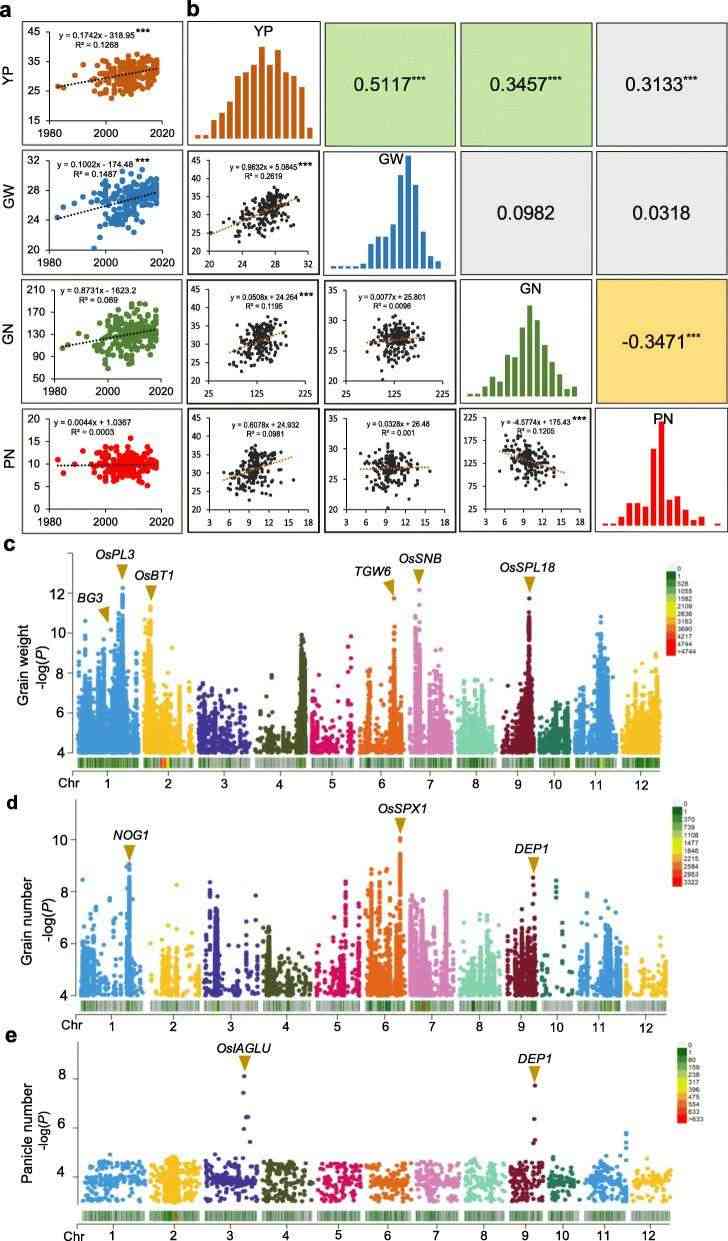 GWAS of genes related to yield traits (Xiao et al., 2021)
GWAS of genes related to yield traits (Xiao et al., 2021)