DNA Methylation Array Development: Evolution, Innovation, and Future Trends
DNA methylation, a critical modification within the field of epigenetics, plays a pivotal role in regulating gene expression, cell differentiation, development, and the onset of various diseases. Under normal physiological conditions, the methylation level of DNA is essential for maintaining genomic stability. However, in diseases such as cancer and aging, aberrant DNA methylation frequently constitutes a pivotal factor leading to gene dysfunction and disease development. In recent years, significant advancements have been made in the study of DNA methylation, particularly in the identification of disease markers and the application of DNA methylation analysis in disease diagnosis, due to the development of high-throughput sequencing technology and bioinformatics.
A notable advancement in this field is the development of DNA methylation array technology, which enables comprehensive analysis of methylation status across the genome. Since the introduction of Illumina's Infinium technology, methylation arrays have become integral tools in epigenomics research. These arrays facilitate efficient detection of methylation at thousands of CpG sites, aiding in the early diagnosis of tumors, aging-related conditions, and immune disorders. Continuous optimization and innovation have enhanced data analysis, signal detection, and standardization processes, thereby improving the accuracy and reliability of results.
This article reviews the progression of DNA methylation array technology, tracing its evolution from early low-throughput methods to modern high-throughput arrays. It also explores applications in disease diagnosis and biological research. Looking ahead, emerging technologies such as single-cell methylation sequencing and artificial intelligence-driven data analysis are poised to further elevate the role of DNA methylation arrays in epigenetics research, offering new avenues for precision medicine and personalized treatment strategies.
Evolution of DNA Methylation Arrays
DNA methylation, a fundamental mechanism in epigenetics, has significantly transformed life science research through advancements in detection technologies. From initial explorations of chemical modifications to sophisticated high-throughput microarray technologies, the field's evolution reflects a deepening understanding of gene expression regulation. The advent of methylation microarrays addressed limitations of traditional methods, such as low throughput and limited resolution, facilitating the transition of epigenetics from basic research to clinical applications through standardized and scalable detection systems. This section systematically traces the technological evolution, analyzing key breakthroughs and acknowledging pioneers who have driven innovation.
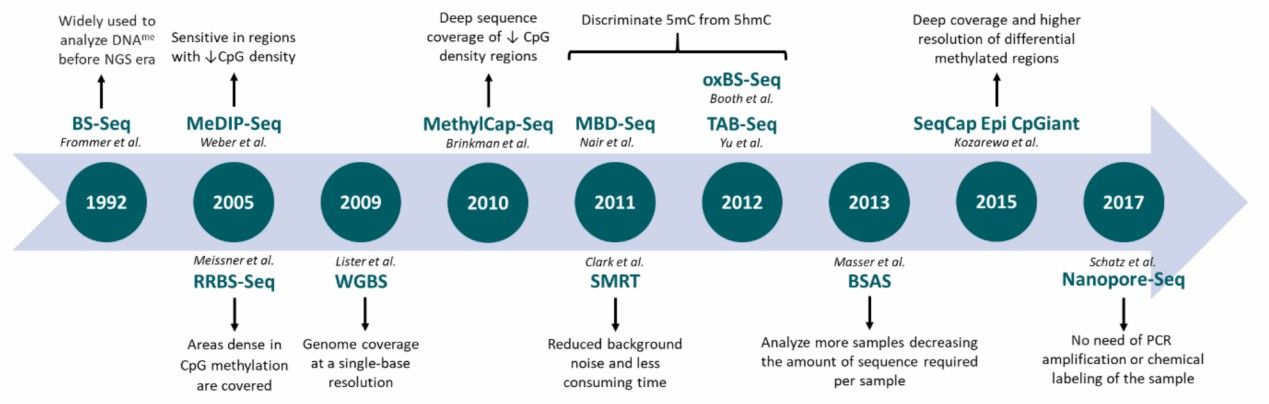 Evolution of next-generation sequencing-based techniques applied to DNA methylation profiling (Barros-Silva et al., 2018)
Evolution of next-generation sequencing-based techniques applied to DNA methylation profiling (Barros-Silva et al., 2018)
Technological Milestones in the Evolution
The journey into DNA methylation research began in 1925 with the identification of 5-methylcytosine (5mC) in Mycobacterium tuberculosis, highlighting the significance of chemical DNA modifications. In the 1950s, the methylation pattern of CpG dinucleotides in mammals was confirmed, revealing methylation's role in gene silencing and genomic imprinting. However, early studies faced technological constraints, relying on low-throughput methods like Southern blotting, which limited resolution to specific sites and hindered genome-wide pattern analysis.
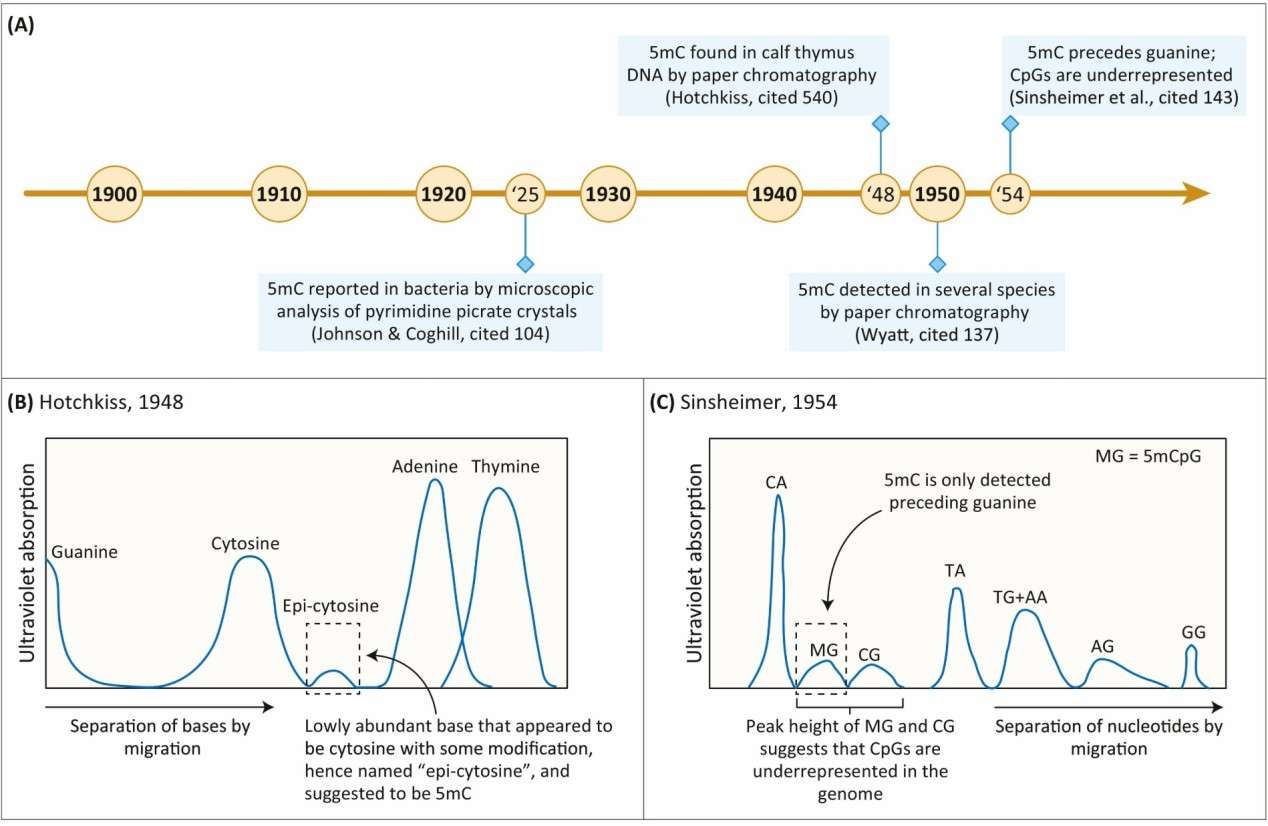 The discovery of 5-methylcytosine (Mattei et al., 2022)
The discovery of 5-methylcytosine (Mattei et al., 2022)
A transformative period emerged in the 1990s with the introduction of gene chip technology, offering new approaches to methylation analysis. In 2000, Eads' team developed MethyLight technology, combining fluorescence quantitative PCR with bisulfite treatment to detect hundreds of loci in parallel. This breakthrough laid the groundwork for subsequent chip designs. Post-2005, the maturation of second-generation sequencing technologies demanded single-base resolution detection, accelerating chip technology iterations. In 2009, Illumina commercialized the technology with the 27K methylation chip, marking its application phase. Subsequent releases of the 450K and 850K microarrays expanded coverage to include enhancers, open chromatin, and other regulatory elements through a hybrid design integrating type I and type II probes. This integration reduced sample requirements to 2 μg of DNA, significantly enhancing clinical utility.
Recent years have witnessed rapid advancements in deep learning and multi-omics integration, reshaping technological boundaries. For instance, the DeepMethylation framework employs word embedding techniques to analyze DNA sequence features, achieving methylation site prediction accuracy exceeding 97.8%. Additionally, integrating nanopore sequencing with single-cell analysis propels methylation detection towards real-time dynamic resolution and cellular heterogeneity research.
Pioneering Studies and Influential Contributors
Several scientists and teams have been instrumental in advancing DNA methylation array technology. Illumina, a leading industry player, addressed single-nucleotide resolution detection challenges by chemically modifying methylated sites through its Infinium technology platform. The progression from 27,000 to 935,000 probes represents a 30-fold increase in site coverage, significantly broadening clinical research applications. This enhancement was achieved by optimizing probe designs to ensure compatibility with formalin-fixed samples.
Chinese researchers have also made significant contributions. Professor Zhu Jingde's team led the "Epigenetic Star Map Project," constructing a 100,000-person methylome database to reveal tissue-specific tumor methylation patterns. Their proposed liquid biopsy technology demonstrated over 90% sensitivity in early cancer diagnosis. Separately, Kong Qingpeng's team at the Chinese Academy of Sciences identified male-specific longevity-related differentially methylated regions through whole-genome methylation sequencing, offering new insights into epigenetic aging research.
The DeepMethylation team further enhanced methylation site prediction models using deep learning algorithms, improving non-experimental data prediction accuracy and supporting intelligent chip design. Concurrent studies, such as the mammalian methylation map by Cokus et al. and cancer methylation regulatory targets identified by Yang et al., have provided critical references for microarray probe selection and disease target screening. Collectively, these pioneering efforts bridge the gap between fundamental discoveries and practical clinical applications.
Service you may intersted in
Learn More:
- DNA Methylation Arrays: Principles, Technologies, and Applications
- DNA Methylation Array Principles: Core Mechanisms and Technological Insights
- DNA Methylation Array Workflow & Analysis: Process Optimization and Data Insights
- DNA Methylation Array Applications: Bridging Clinical Breakthroughs and Research Innovations
Technological Innovations
The progression of DNA methylation microarray technology has transitioned from initial exploratory tools to sophisticated platforms enabling precise epigenetic profiling. Early microarrays, while groundbreaking, faced limitations in coverage and signal accuracy, restricting comprehensive epigenetic studies. The introduction of Illumina's Infinium platform marked a significant advancement, integrating improvements in probe chemistry, signal detection, and data analysis. These enhancements addressed previous challenges and ushered in an era of single-nucleotide resolution and genome-wide coverage, laying a robust foundation for disease mechanism exploration and clinical research.
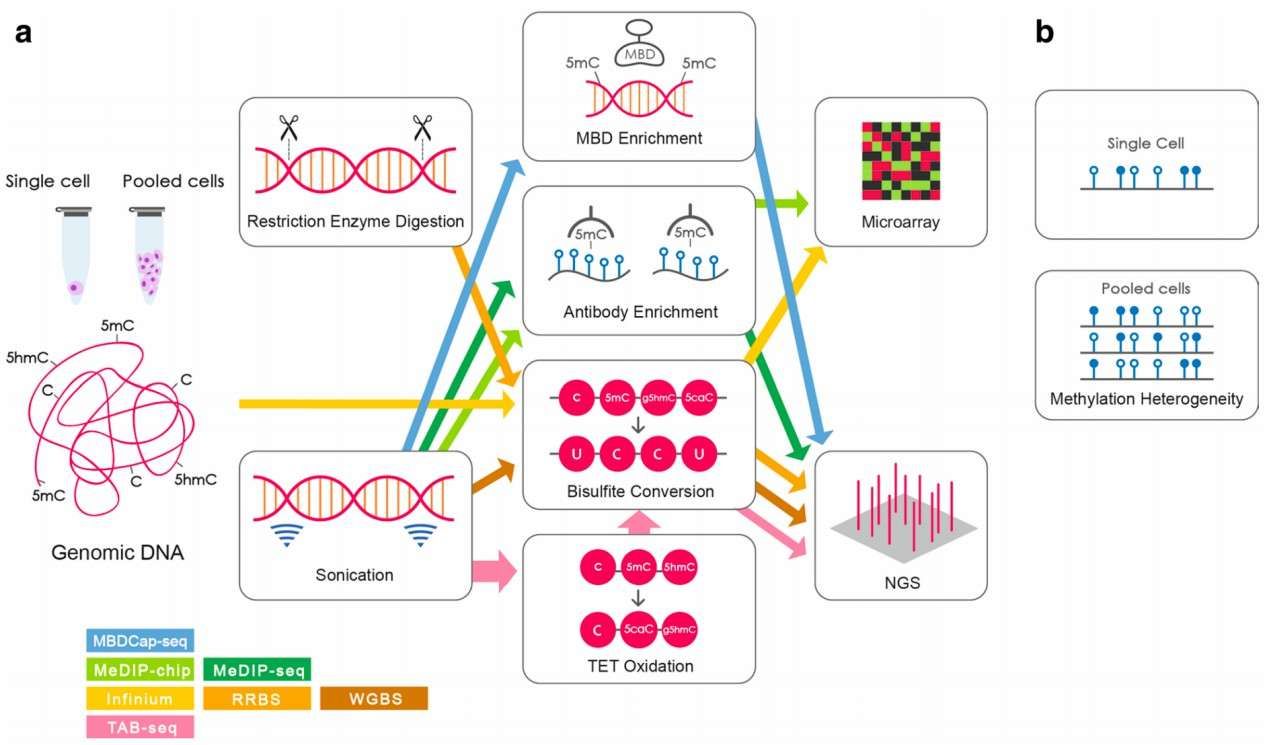 Commonly used methods for genome-wide DNA methylation analysis (Yong et al., 2016)
Commonly used methods for genome-wide DNA methylation analysis (Yong et al., 2016)
From Early Microarrays to Illumina Infinium
Initial DNA methylation microarrays, such as the Illumina GoldenGate BeadArray, utilized bisulfite treatment to differentiate between methylated and unmethylated cytosines, followed by a dual-probe hybridization approach. However, these early platforms were limited in scope, primarily focusing on CpG islands in promoter regions, and were susceptible to probe mismatches due to bisulfite-induced base changes. Additionally, their low throughput and high costs hindered large-scale studies.
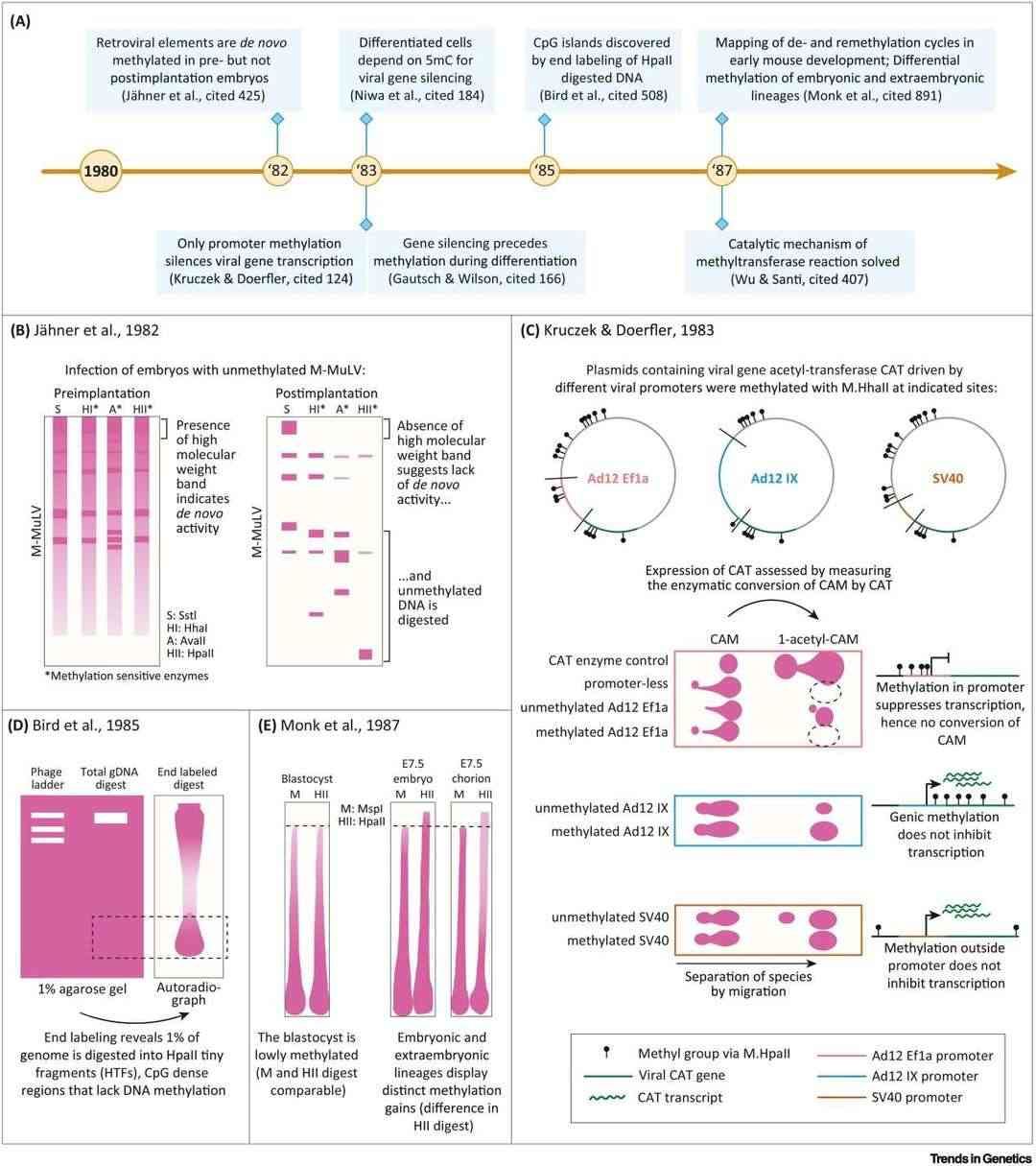 Promoter methylation and the discovery of CpG islands (Mattei et al., 2022)
Promoter methylation and the discovery of CpG islands (Mattei et al., 2022)
To overcome these limitations, Illumina developed the Infinium HumanMethylation450K and EPIC platforms, significantly expanding the number of interrogated CpG sites. The 450K array covered over 480,000 CpG sites, including promoters, gene bodies, and non-coding regions. The subsequent EPIC platform further increased coverage to approximately 935,000 CpG sites, with enhanced representation of enhancer regions, making it particularly suitable for studying complex regulatory networks. Moreover, the Infinium platform demonstrated high compatibility with various sample types, including formalin-fixed, paraffin-embedded (FFPE) tissues, requiring as little as 1 µg of DNA. The integration of the automated iScan imaging system and standardized analysis tools like GenomeStudio contributed to the widespread adoption of the Infinium platform in methylation research.
Technological Innovations in Probe Design
Advancements in probe design have been central to the technological progress of the Infinium platform. Earlier microarrays required separate probes for methylated and unmethylated states at each CpG site, leading to increased chip space usage and signal bias due to differences in hybridization efficiency. The Infinium platform addressed these issues by implementing a hybrid design of Type I and Type II probes. Type I probes, utilizing a two-probe strategy, effectively target hypermethylated regions such as promoter CpG islands, ensuring precise detection of key regulatory sites. Type II probes employ Single Base Extension (SBE) technology, allowing a single probe to differentiate methylation status by incorporating fluorescently labeled nucleotides, thereby increasing chip density and coverage.
Simultaneously, signal detection techniques were optimized to enhance data reliability. Previous platforms used distinct fluorescent dyes for labeling, which were prone to background noise. The Infinium platform improved signal capture by refining probe design at the 3' end and incorporating high-resolution in silico bead arrays with the iScan system. The introduction of the dynamic signal range extension algorithm effectively corrected biases in extreme methylation values. Additionally, data analysis algorithms like Beta Mixture Quantile (BMIQ) and Subset-quantile Within Array Normalization (SWAN) were developed to reduce systematic errors and optimize the effect of GC content on signal intensity, respectively.
Performance Optimization Strategies
DNA methylation microarrays are essential tools in epigenetic research, directly impacting the accuracy of disease marker identification and molecular mechanism analysis. As the demand for high-throughput assays grows, researchers face challenges in balancing assay sensitivity and specificity, mitigating batch effects, and enhancing data reproducibility. Recent advancements, including innovative probe designs, advanced computational correction algorithms, and standardized processes, have addressed these challenges, establishing a solid foundation for multi-center cohort studies and clinical applications.
Enhancing Sensitivity and Specificity
Improving the sensitivity and specificity of microarray detection involves optimizing probe coverage, controlling non-specific signals, and implementing highly sensitive detection platforms. The latest platforms, such as the Illumina EPIC chips, have increased probe density in functional regions by expanding CpG site coverage to include regulatory elements, enhancers, and conserved regions. Excluding probes with high homology to sex chromosome sequences helps reduce false positives from cross-hybridization.
To enhance signal detection, combining ligase cycling reaction (LCR) or dendritic DNA signal amplification technologies has proven effective. These methods enable the detection of methylation signals at single-base resolution, even at very low concentrations, increasing sensitivity to the femtomolar level. Integrating microfluidic and cellular platforms is advancing the development of portable assays that operate without instruments, broadening their applicability. For specificity control, techniques like MethyLight and SMART-MSP improve quantification through fluorescent probes or melting curves, effectively avoiding errors from incomplete bisulfite conversion. Functional normalization methods distinguish between technical and biological variations, ensuring the true signal remains unaltered.
Mitigating Batch Effects and Improving Reproducibility
Batch effects significantly influence the reliability of methylation data, particularly in multicenter or long-term projects. To prevent technical biases from affecting biological interpretations, a comprehensive control system is essential, encompassing everything from sample collection to data analysis. In experimental design, systematically distributing samples across different batches and standardizing DNA extraction and hybridization processes can reduce systematic bias.
For data correction, tools like BEclear and ComBat-met, calibrated according to latent factor models or Beta regression, enhance statistical efficacy while preserving genuine signals. When combined with RUVm and functional normalization, these methods effectively ensure data consistency in large-scale samples. Principal component analysis (PCA) and the Kolmogorov-Smirnov test can assess batch effects by identifying abnormal patterns and assisting in the elimination of outlier samples, thereby improving overall data quality and reproducibility.
Future Perspectives
As epigenetic research advances, DNA methylation microarrays are evolving into technologies with increased throughput, stronger integration, and broader adaptability. The future integration of these platforms with next-generation sequencing (NGS) technologies is expected to reshape research strategies. For example, using high-throughput microarrays for preliminary screening to quickly identify crucial methylated regions, followed by whole-genome resequencing (e.g., WGBS) for precise localization, can enhance the efficacy and depth of subsequent analyses. Additionally, multi-omics integration strategies will combine methylation data with transcriptomic, epigenomic, and metabolomic information, transforming static marker identification into dynamic regulatory mechanism analysis, thus providing comprehensive support for complex disease research and precision medicine.
The incorporation of artificial intelligence and machine learning is revitalizing the analysis and application of DNA methylation microarrays. Currently, deep learning models assist in CpG site screening, feature extraction, and disease classification prediction, demonstrating remarkable proficiency in managing high-dimensional, non-linear epigenomic data. With ongoing algorithm optimization, the development of end-to-end automated analysis processes-from raw signal processing to biological interpretation-is anticipated, significantly improving data utilization efficiency and clinical translation potential. Furthermore, exploring AI-driven probe optimization design holds promise in achieving two key objectives: capturing more critical biological information with fewer probes and advancing the chip platform into intelligent processing.
References
- Mattei, Alexandra L et al. "DNA methylation: a historical perspective." Trends in genetics : TIG vol. 38,7 (2022): 676-707. doi:10.1016/j.tig.2022.03.010
- Barros-Silva, Daniela et al. "Profiling DNA Methylation Based on Next-Generation Sequencing Approaches: New Insights and Clinical Applications." Genes vol. 9,9 429. 23 Aug. 2018, doi:10.3390/genes9090429
- Yong, Wai-Shin et al. "Profiling genome-wide DNA methylation." Epigenetics & chromatin vol. 9 26. 29 Jun. 2016, doi:10.1186/s13072-016-0075-3




