DNA Methylation Array Workflow & Analysis: Process Optimization and Data Insights
As a foundational technology for epigenetic research, the optimization of standardized processes and data analysis strategies is directly related to the depth of disease mechanism analysis and the feasibility of clinical translation. In the context of emerging technologies, such as single-cell epigenomics, liquid biopsy, and other cutting-edge approaches, the ability to achieve high-fidelity detection in complex samples, including FFPE tissues and circulating free DNA, has emerged as a pivotal challenge in technological refinement. This paper systematically examines the entire process of standardized operations, from sample preparation and bisulfite conversion to chip hybridization, with a focus on innovative solutions for quality control nodes. For instance, to address the DNA breakage problem caused by traditional transformation technology, novel enzyme transformation technology (e.g., EM-seq) reduces the degradation rate to less than 5%. At the level of data analysis, AI-based quality control tools (e.g., MethylAid) automate the identification of abnormal samples through multi-dimensional feature clustering. These technological breakthroughs not only enhance the resolution accuracy of methylation profiles, but also lay the foundation for precision medicine in the era of multi-omics integration.
This paper explores the optimization path of DNA methylation microarrays in all aspects, from experimental manipulation to bioinformatics analysis. By conducting a thorough analysis of critical quality control indicators during sample preparation, molecular engineering strategies for optimizing hybridization conditions, and machine learning-driven frameworks for conducting differential methylation analysis, this study establishes a replicable technical paradigm for researchers in the field. Additionally, the article provides insights into translational cases of multi-omics integration in cancer and neurodegenerative diseases, revealing how methylation regulatory networks link clinical phenotypes across molecular levels. The potential value of methylation data in mechanistic interventions and targeted therapies is demonstrated in cases such as BRAF mutation-induced episodic silencing in thyroid cancer and spatiotemporal co-localization of the SORL1 gene in Alzheimer's disease.
Service you may intersted in
Learn More:
- DNA Methylation Arrays: Principles, Technologies, and Applications
- DNA Methylation Array Development: Evolution, Innovation, and Future Trends
- DNA Methylation Array Principles: Core Mechanisms and Technological Insights
- DNA Methylation Array Applications: Bridging Clinical Breakthroughs and Research Innovations
Step-by-Step Workflow
As the foundational methodology of epigenetic research, the reliability of DNA methylation microarray technology is contingent upon the standardization and refinement of the experimental process. From the moment a sample is collected to the point at which data is generated, minor inconsistencies in each stage of the process have the potential to compromise the precision of methylation profiles. In the context of emerging technologies such as single-cell epigenomics and liquid biopsy, achieving high-fidelity detection in complex samples (e.g., FFPE tissues or circulating free DNA) has emerged as a pivotal challenge for technological optimization. In this section, we will systematically analyze the standardized operation procedure of methylation microarrays and focus on the key quality control nodes to provide researchers with replicable experimental paradigms.
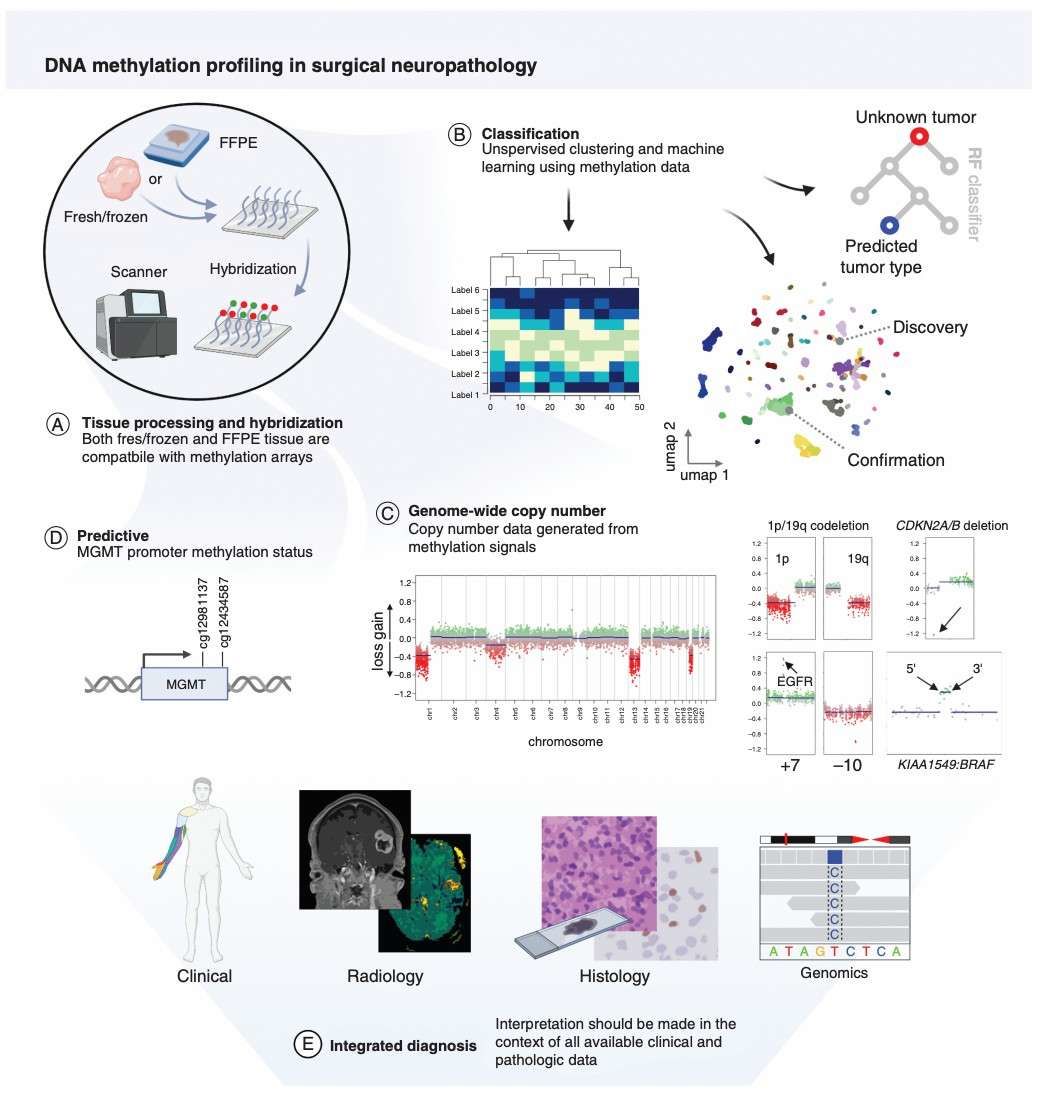 Workflow and applications of DNA methylation profiling (Pratt et al., 2021)
Workflow and applications of DNA methylation profiling (Pratt et al., 2021)
Sample preparation
The quality of the sample preparation stage is directly correlated with the confidence of the subsequent data obtained from methylation microarray analysis. For fresh or frozen tissues, high-purity DNA (A260/A280 ratio 1.8-2.0) should be extracted by phenol-chloroform or magnetic bead extraction. For FFPE samples, cross-linking damage should be repaired by dewaxing and proteinase K digestion, combined with fragment screening (e.g., AMPure XP magnetic beads) to remove short-strand degradation products. In recent years, automated microfluidic chip-based extraction systems (e.g., QIAcube) have significantly improved the DNA recovery rate of FFPE samples (up to 80% or more), providing a technological safeguard for clinical retrospective studies.
The subsequent step involves the core process of sulfite conversion, where unmethylated cytosine (C) is transformed into uracil (U) under acidic conditions, while methylated C remains unaltered. The conversion efficiency, which is typically required to be greater than 95%, can be monitored using spike-in controls, such as Lambda DNA. It is noteworthy that conventional sulfite treatment is susceptible to DNA breakage, with a degradation rate of ~30-50%. In contrast, novel enzymatic transformation techniques, such as EM-seq, exhibit a significantly reduced degradation rate of less than 5%, attributable to their mild reaction conditions, which renders them particularly well-suited for the processing of micro samples. Subsequent to the transformation process, the DNA must undergo a purification and amplification step to ensure its suitability for hybridization with microarray probes. Strict quality control measures during sample preparation are imperative for the construction of high-resolution methylation profiles.
Chip hybridization and signal capture
Hybridization is the "signal amplifier" of methylation detection, and its condition optimization directly affects the signal-to-noise ratio of data. Utilizing the Illumina Infinium platform as a case study, the transformed single-stranded DNA and the chip probe (50-70 bp) are amalgamated through base complementarity pairing. In this scenario, the type I probe discerns the methylation status through two-color fluorescence (Cy3/Cy5), while the type II probe facilitates high-throughput detection through a probe-based strategy with multiple labels. The salt ion concentration of the hybridization buffer (typically 3M TMAC) and the temperature gradient (45-55°C) must be meticulously regulated to balance the probe binding force with the background signal.
In recent years, molecular engineering techniques (e.g., probe shielding) have been employed to reduce background noise by more than 30% by introducing blocking sequences to inhibit non-specific binding. Subsequent to hybridization, the chip undergoes rigorous elution (e.g., low-salt SSC buffer) to expel unbound DNA, and then the fluorescence signal is captured by a high-resolution scanner (e.g., iScan system). For the purpose of signal extraction, the calculation of β-value (β=methylation signal/total signal) must be corrected for fluorescence intensity bias, and Illumina's GenomeStudio software automatically filters low-quality probes through built-in quality control modules (e.g., detecting p-value < 0.01). The optimization of hybridization conditions and the automation of signal resolution are pivotal in enhancing data consistency.
Quality Control and Troubleshooting
The quality control system is implemented at each stage of the methylation microarray process. At the sample level, DNA integrity (DV200>50%), transformation efficiency (quantified by spike-in), and fragment distribution (Agilent Bioanalyzer) are assessed. At the hybridization stage, built-in quality control probes (e.g., staining control probes) allow for real-time monitoring of the batch effect; prior to data analysis, inter-array differences need to be eliminated through quartile. Prior to data analysis, quartiles are standardized to eliminate inter-chip variation, and probe type bias is corrected using the BMIQ algorithm (Infinium I/II).
Common problems, such as low signal intensity, can be mitigated by extending hybridization time (16-24 hours) or increasing the amount of DNA input (500 ng or more); in the case of batch effects, the ComBat algorithm effectively corrects for technical variability. Currently, artificial intelligence (AI)-driven quality control (QC) tools (e.g., MethylAid) have automated the identification of abnormal samples, thereby reducing the false-negative rate to less than 5% through multidimensional feature clustering. QC functions not only as a data filtration mechanism but also as a foundational principle for experimental design.
Advanced Data Analysis Pipeline
The substantial volume of data produced by DNA methylation microarrays represents a valuable resource for epigenetic studies and a significant challenge for computational biology. The intricate interplay of technical noise, such as microarray batch effects and probe design biases, with biological variation, including cellular heterogeneity, within the raw signals, necessitates the implementation of data normalization and differential analysis techniques to effectively resolve methylation profiles. The advent of single-cell epigenomics and multi-omics integration has led to a paradigm shift in the traditional analysis process, moving from "batch correction" to "intelligent deconvolution." This session will focus on the standardization strategy and differential analysis framework of methylation data, and will explore how cutting-edge bioinformatics tools can enable precise epigenetic decoding.
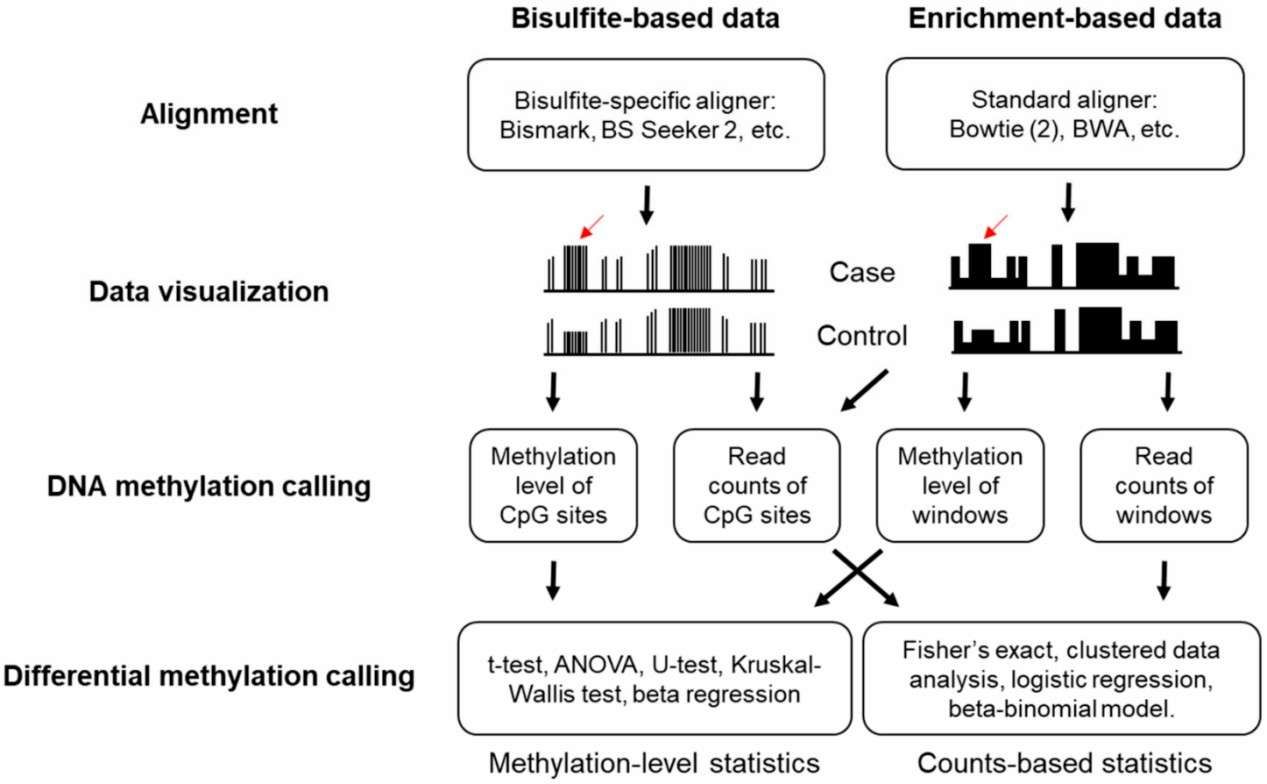 Computational pipeline for DNA methylation sequencing data analysis (Huang et al., 2019)
Computational pipeline for DNA methylation sequencing data analysis (Huang et al., 2019)
Data standardization and differential methylation analysis
Raw methylation data, characterized by β-values or M-values, is subjected to multiple levels of correction to enhance comparability. Quantile normalization, a classical method, has been shown to effectively eliminate systematic bias between chips by enforcing consistent signal distribution across samples. However, disparities in hybridization efficiency between type I and type II probes (e.g., elevated signal fluctuations for type II probes) within the Illumina Infinium platform necessitate additional processing.The BMIQ (Beta Mixture Quantile dilation) algorithm facilitates cross-platform data integration by constructing a hybrid model to rectify the distribution bias of the two probe types separately.
The crux of differential methylation analysis entails the identification of CpG sites (DMPs) or regions (DMRs) that exhibit significant alterations in methylation levels between groups. Linear regression models (e.g., limma) and Bayesian methods (e.g., Bumphunter) are frequently employed to adjust for confounders (e.g., age, gender). To address the issue of multiple testing, the Benjamini-Hochberg correction can maintain a false positive rate below 5%. In recent years, the machine learning-based WEN (Weighted Elastic Net) model has achieved highly sensitive detection of low-abundance methylation signals (<1%) in early cancer screening by integrating spatial correlations of CpG sites. The core challenge in discerning technical noise from authentic biological signals lies in the synergistic optimization of normalization and difference analysis.
Bioinformatics pipeline
The analysis of methylation data is highly dependent on the computational ecosystem that is built by specialized tools. The Bioconductor community's minfi package offers a comprehensive solution that encompasses raw data import (IDAT file), QC, and standardization. The package's integrated dmrcate module automatically identifies DMRs and annotates regulatory elements in conjunction with the ENCODE database. For clinical researchers, graphical tools such as MethylAid provide interactive QC visualization, while Illumina's GenomeStudio supports "one-click" report generation, significantly lowering the threshold of analysis.
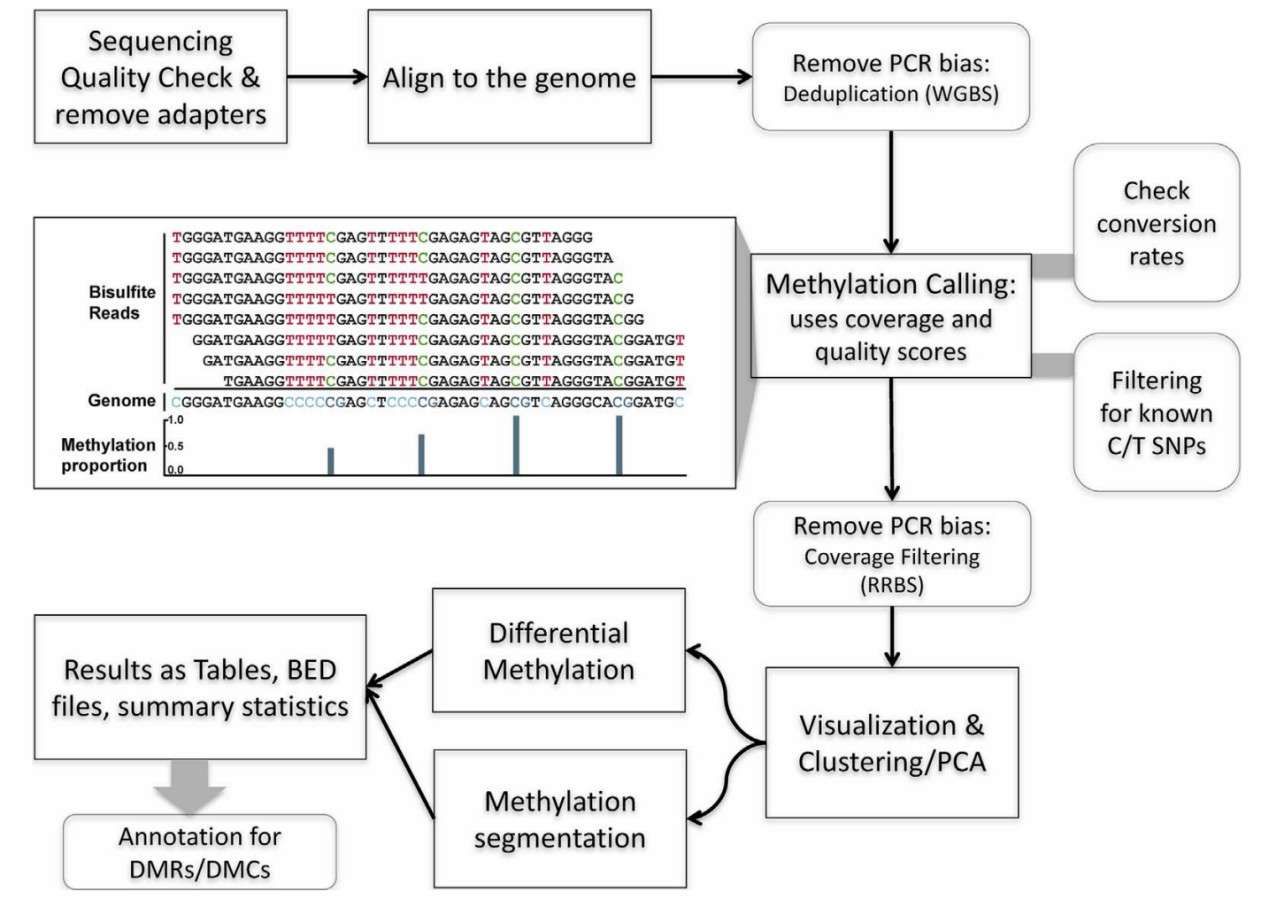 Workflow for analysis of DNA methylation using data from bisulfite sequencing experiment (Wreczycka et al., 2017)
Workflow for analysis of DNA methylation using data from bisulfite sequencing experiment (Wreczycka et al., 2017)
At the level of mechanism mining, the tool chain is extending to multi-omics integration and spatio-temporal dynamic analysis. For instance, ChAMP (Chip Analysis Methylation Pipeline) can construct gene regulatory networks by correlating methylation data with transcriptome/epigenetic modifications (e.g., ATAC-seq). In the context of single-cell methylation data, MIRA (Methylation Integration and Regulatory Analysis) employs graph neural networks to identify cell subpopulation-specific epigenetic programs. Furthermore, cloud platforms, such as Google MethylFlow, are beginning to offer automated, AI-based analysis services with pre-trained models that can complete DMP screening of thousands of data points in less than 10 minutes. The modularity and intelligence of bioinformatics tools are transforming methylation data into actionable biological knowledge.
Integrative Approaches in Multi-Omics
In the current era of multi-omics, the interpretation of DNA methylation microarray data has evolved beyond the confines of epigenetic changes in a single dimension. Instead, it now unveils the global regulatory logic of biological processes through the integration with gene expression, protein interactions, and other multi-level data. The crux of this integration strategy lies in transcending the limitations of conventional association analysis, situating methylation modifications within a dynamic regulatory network, and examining their interactions with transcriptional activities, signaling pathways, and other phenomena. A synergistic analysis of methylation and gene expression, for instance, necessitates the utilization of standardized tools (e.g., minfi) to eliminate technical bias, in addition to leveraging causal inference models to discern direct regulatory effects from indirect associations. The advent of single-cell sequencing and spatial multi-omics technologies has empowered researchers to observe the spatial and temporal interplay of methylation and expression in specific cellular subpopulations, offering novel insights into the analysis of disease heterogeneity.
Recent research has achieved success in predicting gene activity and identifying key regulatory nodes through the integration of single-cell methylation and transcriptome data using machine learning frameworks such as MAPLE. A case study of cancer research demonstrates the efficacy of this approach. For instance, the methylation-expression co-regulation module of the RNASET2 gene in lung cancer has been found to be closely associated with tumor microenvironment remodeling. Similarly, studies on thyroid cancer have revealed that the BRAF V600E mutation drives the sustained activation of the MAPK pathway by inducing hypermethylation of the DUSP4 promoter. In the domain of neurodegenerative diseases, an integrated multi-omics analysis of Alzheimer's disease confirmed the spatial co-localization of episodic silencing of the SORL1 gene with β-amyloid deposition, highlighting the causal role of methylation regulation in disease progression. These cases demonstrate that cross-omics associations can transcend the molecular divide, thereby establishing a direct link between epigenetic variants and clinical phenotypes. This development provides a theoretical foundation for the development of targeted therapies.
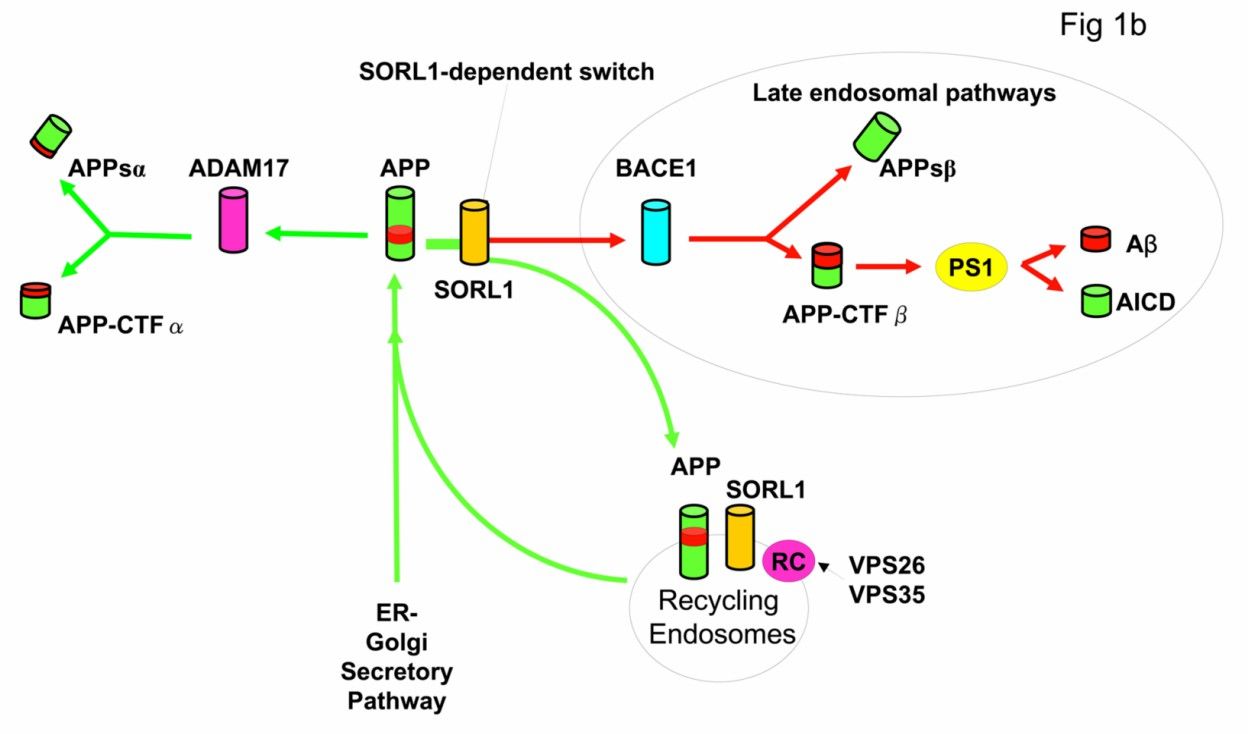 Diagram of the amyloid precursor protein processing pathways (Rogaeva et al., 2007)
Diagram of the amyloid precursor protein processing pathways (Rogaeva et al., 2007)
The optimization of data standardization processes and the iteration of artificial intelligence algorithms have contributed to the acceleration of multi-omics integration from basic research to clinical translation. In the future, the combination of spatial epigenomics and real-time dynamic tracking technology will enable researchers to more accurately analyze the spatial and temporal specificity of methylation regulation. This, in turn, will promote a transition from "association discovery" to "mechanism intervention" in precision medicine. Achieving this breakthrough necessitates not only technological innovation but also the establishment of an interdisciplinary collaborative framework, thereby facilitating a closed-loop process that encompasses data fusion and disease mechanism analysis.
References
- Huang, Jinyong, and Liang Wang. "Cell-Free DNA Methylation Profiling Analysis-Technologies and Bioinformatics." Cancers vol. 11,11 1741. 6 Nov. 2019, doi:10.3390/cancers11111741
- Wreczycka, Katarzyna et al. "Strategies for analyzing bisulfite sequencing data." Journal of biotechnology vol. 261 (2017): 105-115. doi:10.1016/j.jbiotec.2017.08.007
- Pratt, Drew et al. "DNA methylation profiling as a model for discovery and precision diagnostics in neuro-oncology." Neuro-oncology vol. 23,23 Suppl 5 (2021): S16-S29. doi:10.1093/neuonc/noab143
- Rogaeva, Ekaterina et al. "The neuronal sortilin-related receptor SORL1 is genetically associated with Alzheimer disease." Nature genetics vol. 39,2 (2007): 168-77. doi:10.1038/ng1943






