DNA Methylation Arrays: Principles, Technologies, and Applications
DNA methylation, a fundamental epigenetic mechanism that regulates gene expression, has been linked to various diseases, including cancer and aging. Over the past two decades, methylation microarray technology has undergone a significant paradigm shift, progressing from single-site detection to genome-wide high-precision analysis through the innovative integration of chemical transformation and probe hybridization. Initially, the technology covered only 27,000 CpG sites; however, it has evolved to the point where the EPIC chip now integrates 850,000 functional regions. This technological progression has enhanced not only the throughput and accuracy of detection, but also resolved significant challenges, such as the compatibility of clinical samples and the correction of batch effects through standardized data flow and artificial intelligence algorithms. The technology has now transcended the conventional boundaries of scientific research, demonstrating its potential in fields such as liquid biopsy, single-cell analysis, and real-time nanopore sequencing. This advancement has led to a paradigm shift in epigenetic research, moving from static mapping to spatio-temporal dynamic analysis. Furthermore, it has facilitated a comprehensive technological chain, ranging from mechanism exploration to clinical translation for precision medicine.
In this paper, we systematically analyze the technical principles, optimization strategies, and application prospects of DNA methylation microarrays. The article discusses the translational potential of this technology in early cancer screening, aging clock construction, and environmental health monitoring, and looks forward to the future trend of its integration with single-molecule sequencing and digital health platforms. Readers will thus be provided with a panoramic technological picture from the laboratory to the clinic.
Service you may intersted in
Learn More:
- DNA Methylation Array Development: Evolution, Innovation, and Future Trends
- DNA Methylation Array Principles: Core Mechanisms and Technological Insights
- DNA Methylation Array Workflow & Analysis: Process Optimization and Data Insights
- DNA Methylation Array Applications: Bridging Clinical Breakthroughs and Research Innovations
DNA Methylation Arrays Basics
DNA methylation is a pivotal epigenetic mechanism that regulates gene expression. Its abnormalities are associated with diseases such as cancer and aging. Methylation microarray technology facilitates genome-wide high-throughput detection by integrating bisulfite conversion and probe hybridization. The progression of this technology, from early low-throughput platforms to modern high-precision microarrays, has not only advanced fundamental research in epigenetics but also demonstrated considerable potential in clinical diagnosis. This chapter aims to provide a comprehensive analysis of the fundamental principles, technological evolution, and future directions of this field.
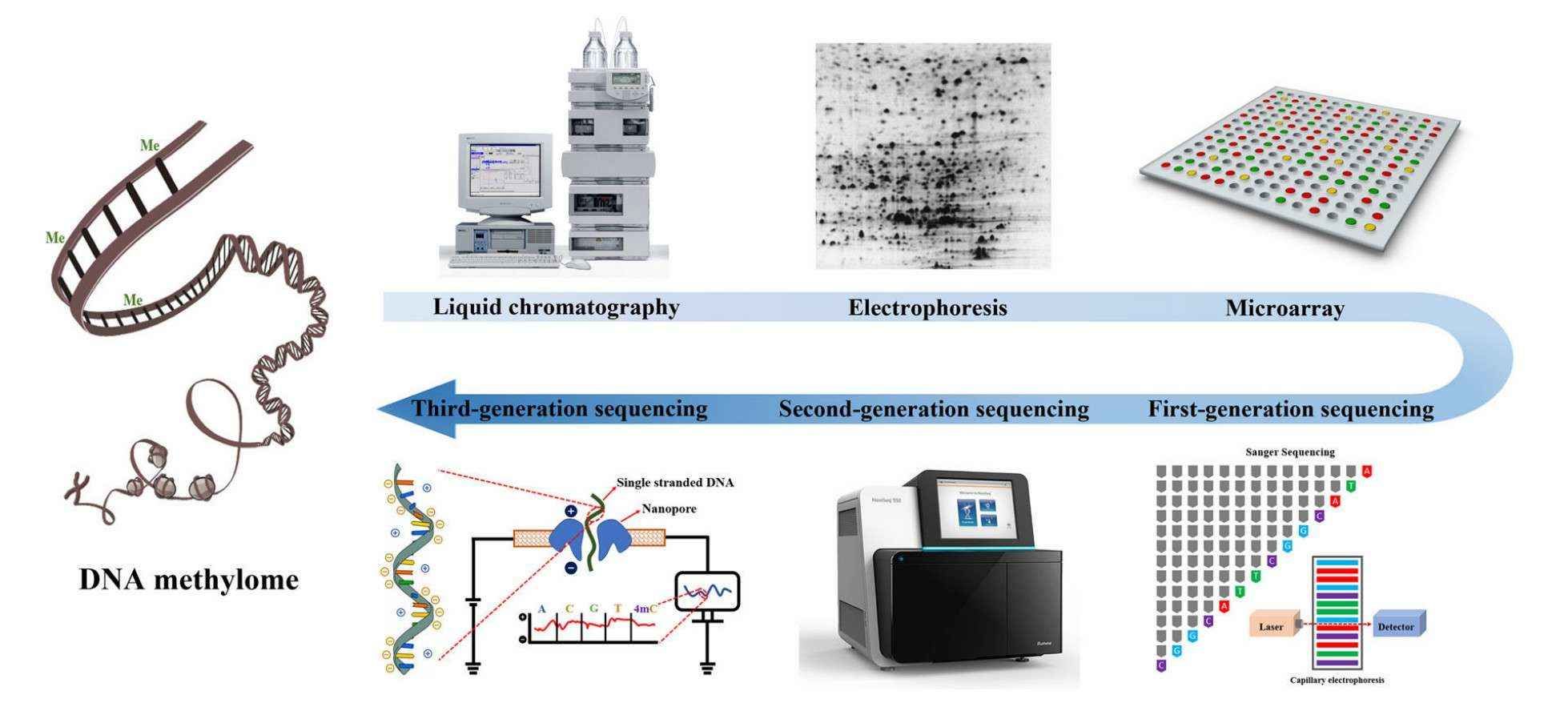 The evolution of DNA methylation profiling methodology (Li et al., 2021)
The evolution of DNA methylation profiling methodology (Li et al., 2021)
Detection Principle: Chemical Conversion and Probe Design
The methylation assay is predicated on the distinction between methylated (5mC) and unmethylated cytosine. Bisulfite treatment deamidates unmethylated cytosine, resulting in the conversion to uracil, while the methylation site remains unaltered. The design of microarray probes was tailored for the transformed sequences, and the quantification of methylation levels was performed by measuring the hybridization signal intensity. The Illumina Infinium platform, for instance, utilizes single base extension (SBE) technology with a two-color fluorescence system to detect methylation status at single nucleotide resolution.
Optimization of probe design has been shown to significantly improve detection performance. While earlier chips (e.g., 27K) only covered CpG islands in the promoter region, the modern 850K EPIC chip extends the detection range to enhancers, open chromatin, and other regulatory regions by mixing probes (Type I/II), and the single probe design of Type II probes increases the density of the chip and reduces the sample requirement (down to 1 μg of DNA) to enhance compatibility with clinical samples (e.g., FFPE) and tissues.
Technological evolution: dual breakthroughs in throughput and accuracy
The development of methylation microarray technology has undergone two significant innovations: throughput enhancement and precision optimization. The 450K microarray of 2009 encompasses 480,000 CpG sites, thereby covering 99% of gene promoters. Building upon this foundation, the 850K EPIC microarray of 2016 further integrates the enhanced sub-database, achieving 90% coverage of functional regions. Concurrent with the enhancement of throughput has been a simultaneous upgrade in signal capture technology. The iScan imaging system, for instance, has reduced the data coefficient of variation (CV) from 15% to 5%. Moreover, the dynamic correction algorithm has addressed the issue of bias in extreme methylation values.
The standardization of data analysis processes has accelerated the adoption of the technology. Tools such as ChAMP and minfi integrate batch effect correction (e.g., ComBat) and functional annotation modules to support the integration of data from multi-center studies. In recent years, deep learning models (e.g., DeepMethyl) have extended the data dimension of microarrays by predicting uncovered sites. Presently, this technology is being integrated with single-cell sequencing and real-time nanopore detection, thereby propelling the field of methylation analysis from static mapping to spatiotemporal dynamic resolution.
DNA Methylation Arrays Workflows
DNA methylation microarrays convert epigenetic information into quantifiable data through a standardized process. The core challenge of this process is balancing detection sensitivity with data reliability. The advent of single-cell technology and liquid biopsy has led to the necessity of maintaining high accuracy in micro-volume samples and enabling cross-platform data calibration. This chapter will therefore analyze the core processes and optimization strategies, and discuss how to promote methylation detection from basic research to clinical translation.
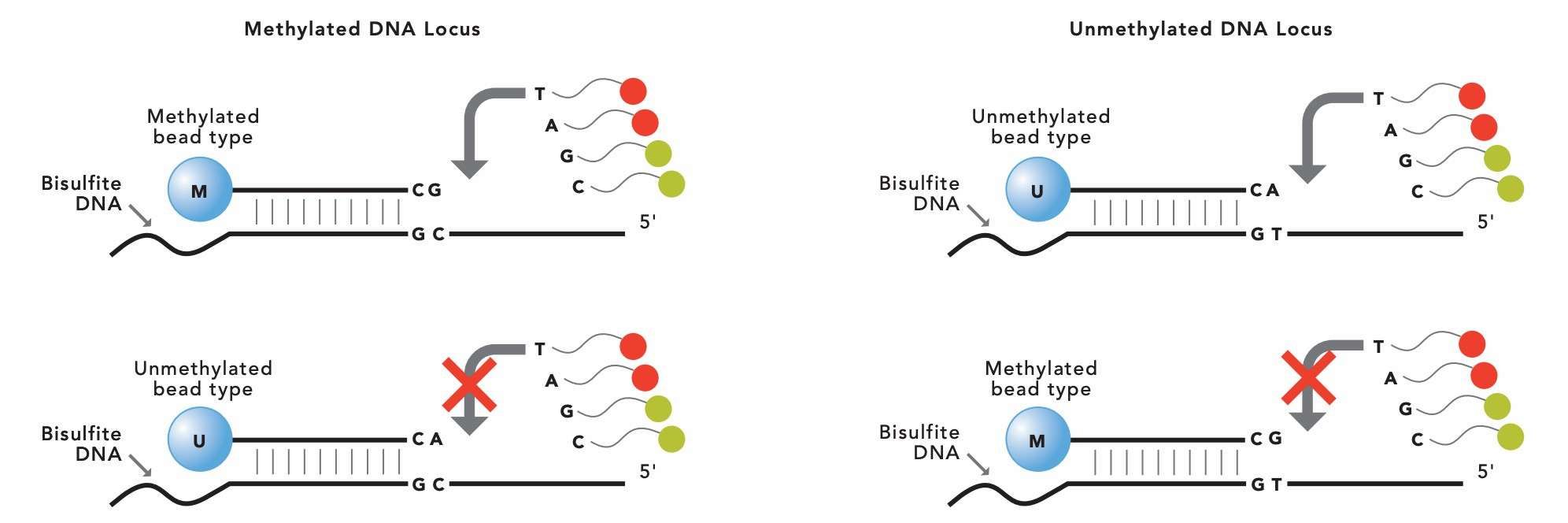 Infinium methylation array experimental principle
Infinium methylation array experimental principle
Standard Process: From Sample Processing to Signal Resolution
The workflow of methylation microarrays commences with the bisulfite treatment of DNA, a step that differentiates between methylated and non-methylated cytosines through chemical transformation. Modern technology utilizes a microfluidic device and antioxidant buffer to increase the conversion efficiency to over 98%, while reducing the rate of DNA degradation and increasing compatibility with low-quality samples. Subsequent to this, the transformed DNA is fragmented and amplified, and then hybridized to the microarray probes. The Illumina platform, for instance, utilizes a two-color fluorescence system (Cy3/Cy5) in conjunction with single-base extension technology, enabling the synchronous detection of hundreds of thousands of CpG sites. These sites encompass promoters, enhancers, and other pivotal regulatory regions.
Following the capture of signals, the methylation level is quantified by β-value and validated with single-base resolution data in combination with targeted sequencing technologies (e.g., TBS-seq). This "microarray-sequencing" strategy offers distinct advantages in the realm of early cancer screening, effectively compensating for the inherent limitations of microarray probe coverage.
Optimization strategies: from probe design to data calibration
The enhancement of detection accuracy is predominantly attributable to probe design and signal optimization. Probe GC content and secondary structure prediction are used to inhibit non-specific binding. One example of this is probe shielding technology, which uses thermodynamic parameters (ΔG values) to block background signals. Microfluidic chips enable dynamic control of hybridization conditions (e.g., salt concentration, temperature), thereby reducing the CV between batches to less than 5% and significantly enhancing experimental reproducibility.
Standardization of data relies on the implementation of sophisticated algorithms and the utilization of reference materials. The SWAN algorithm, for instance, employs quartile correction to mitigate probe type bias, while the NIST methylation reference material (RM 8375) serves as a benchmark for cross-platform data comparison. In the context of liquid biopsies, the fragment length correction model (FLCF) has been instrumental in reducing methylation estimation error from ±8% to ±3% in plasma samples, thereby propelling the technology towards clinical diagnosis.
Service you may intersted in
DNA Methylation Arrays in Data Analysis
The high-throughput nature of DNA methylation microarray data has led to its emergence as a fundamental tool in the field of epigenetic research. However, its analysis faces significant challenges, including the need to address technical noise and the interpretation of biological significance. Contemporary research trends are evolving from unidimensional difference detection to multi-omics dynamic network construction, with a focus on standardizing data, integrating cross-platform information, and elucidating the synergistic regulation of methylation and gene expression. This chapter will focus on data standardization strategies and multi-omics integration pathways, and explore their applications in disease mechanisms and precision medicine.
Data standardization and differential analysis
Raw methylation data (β-values or M-values) necessitate numerous corrections to eliminate technical bias. For instance, in the context of Illumina chips, type I and type II probes must be processed independently due to disparities in hybridization efficiency. The quartile normalization procedure eliminates systematic bias between chips, while the BMIQ algorithm rectifies probe type distribution bias. Machine learning models (e.g., WEN) have been developed to detect low-abundance signals (<1%) in early cancer screening by integrating spatial correlation of CpG sites.
The objective of differential analysis is to identify CpG sites (DMPs) or regions (DMRs) that exhibit significant alterations in methylation levels. Linear regression models (e.g., limma) with Bayesian methods (e.g., Bumphunter) are employed to adjust for confounders such as age and sex, while the Benjamini-Hochberg method is utilized to regulate false-positive rates. At the single-cell level, the variational autoencoder (VAE) addresses the challenge of missing values due to insufficient DNA samples by preserving cellular heterogeneity. The objective of the standardized process is to distinguish technical noise from authentic biological signals, thereby generating high-confidence targets for mechanistic studies.
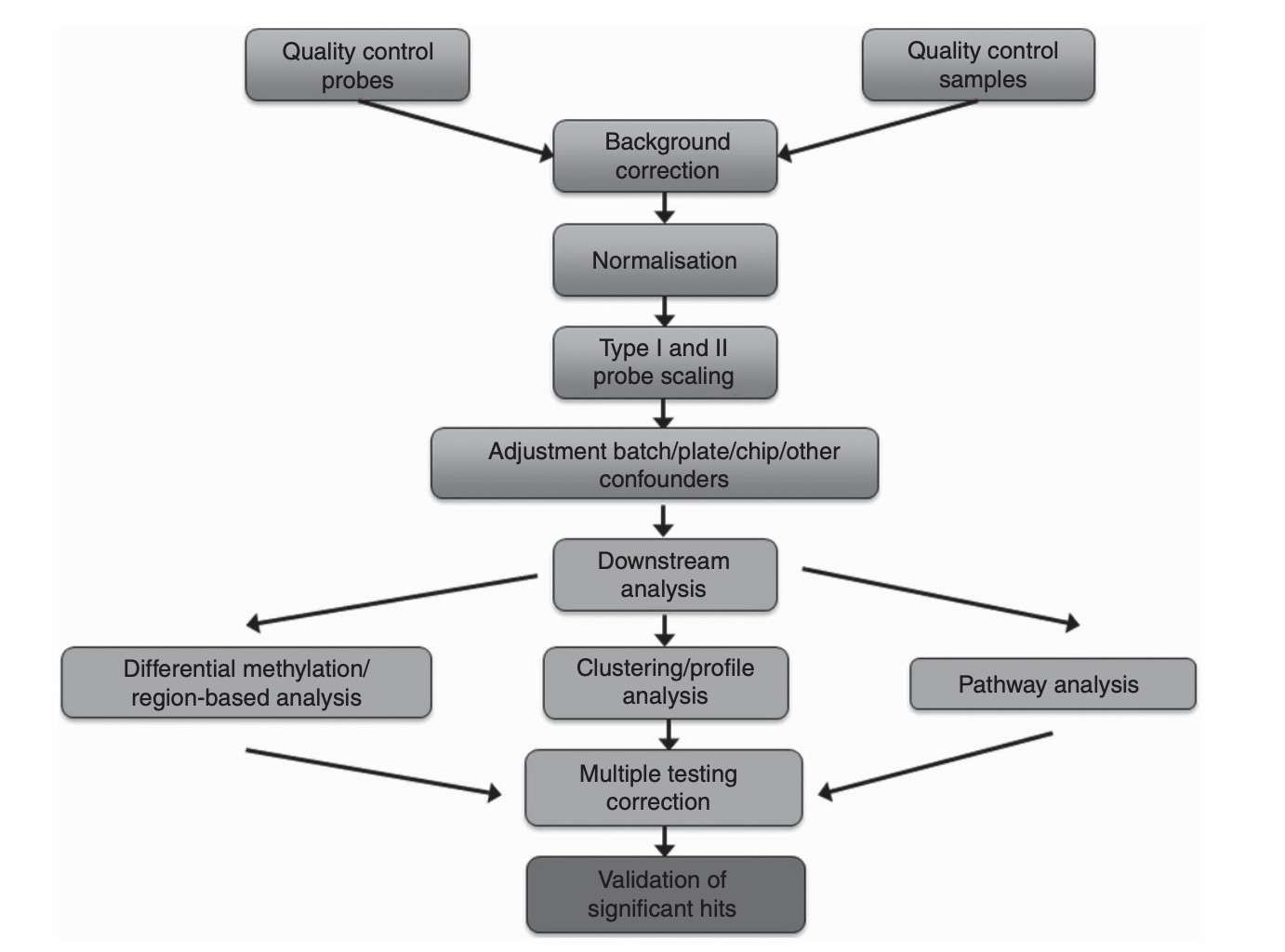 Methylation array data processing and analysis pipeline (Wilhelm-Benartzi et al., 2013)
Methylation array data processing and analysis pipeline (Wilhelm-Benartzi et al., 2013)
Multi-omics integration and dynamic regulation
A comprehensive presentation of the biological significance of methylation data is imperative through multi-omics integration. For instance, the ChAMP process correlates methylation with transcriptome or chromatin accessibility data to construct gene regulatory networks, and the single-cell tool MIRA utilizes graph neural networks to identify cell subpopulation-specific epigenetic programs. Conversely, spatial multi-omics technologies (e.g., DBiT-seq) address the spatial co-localization patterns of methylation and gene expression.
Typically, BRAF mutations in thyroid cancer activate the MAPK pathway by inducing hypermethylation of the DUSP4 promoter; and episodic silencing of the SORL1 gene in Alzheimer's disease is spatiotemporally co-localized with β-amyloid deposition. These findings rely on causal inference models (e.g., MAPLE) to distinguish direct regulatory effects, as well as automated multi-omics analyses on cloud-based platforms (e.g., MethylFlow). The integrated strategy effectively transcends a single molecular dimension, thereby facilitating direct correlation of epigenetic variants with clinical phenotypes. This approach provides a robust theoretical foundation for the development of targeted therapies.
Applications of DNA Methylation Arrays
DNA methylation microarray technology has emerged as a pivotal instrument in the realm of early cancer screening, the exploration of aging mechanisms, and environmental health research. This technology facilitates high-throughput detection of genomic epigenetic modifications, thereby offering a comprehensive and quantitative approach to these research endeavors. The core strength of this technology lies in its ability to transform complex epigenetic signals into quantifiable data, thereby providing a dynamic view of disease diagnosis at the molecular level. The integration of artificial intelligence and multi-omics technologies is propelling methylation analysis from the realm of scientific research to the domain of real-time clinical monitoring. This development is catalyzing the transition of precision medicine to a new era of "predictive intervention."
Clinical translation: early cancer screening and the aging clock
The employment of DNA methylation microarrays has been demonstrated to enhance the sensitivity of cancer diagnosis by facilitating the recognition of tumor-specific epigenetic fingerprints. For instance, in the context of colorectal cancer screening, the SEPT9 gene methylation testing method exhibits a 15% increase in diagnostic accuracy compared to conventional approaches. Similarly, the EpiPanGI Dx system demonstrates the capability to predict six gastrointestinal tumors concurrently with a specificity exceeding 98%. In the context of lung and liver cancer, liquid biopsy technology facilitates non-invasive diagnosis with equivalent tissue biopsy accuracy by tracking methylation markers such as p16INK4A or KCNQ5.
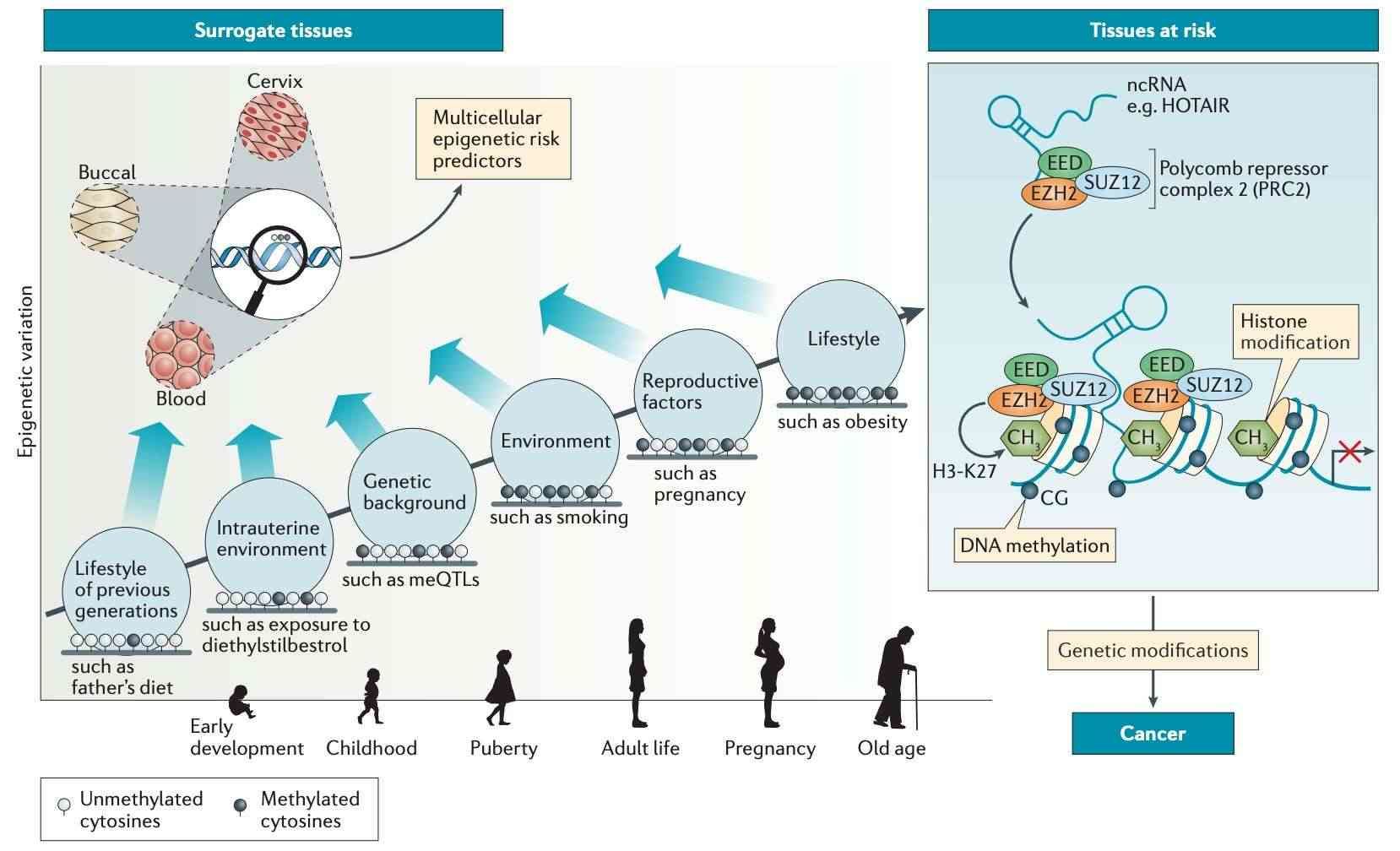 Multicellular predictors of epigenetic risk (Widschwendter et al., 2018)
Multicellular predictors of epigenetic risk (Widschwendter et al., 2018)
In the realm of aging research, methylation clocks (e.g., Horvath clock) have emerged as a means to quantify biological age, thereby facilitating the identification of disease risk. For instance, the methylation age of brain tissue from Alzheimer's disease patients is observed to be 12 years younger than that of blood, while caloric restriction interventions have been shown to reverse the apparent age of mouse liver by up to two years. Furthermore, environmental exposures (e.g., benzene contamination) induce methylation variants that appear earlier than clinical symptoms, providing molecular early warning indicators for occupational health surveillance. These findings underscore the pivotal role of methylation microarrays in elucidating the interplay among environmental factors, aging, and disease pathogenesis.
Technology convergence: AI-driven and multi-omics integration
The field of methylation data analysis is undergoing a paradigm shift due to the integration of artificial intelligence. A deep learning model, DeepSignal, when integrated with nanopore sequencing, has demonstrated the capability to resolve methylation dynamics at the single-molecule level in real time, facilitating effective monitoring of neurodegenerative diseases. Integration of MGMT promoter methylation data into clinical decision-making systems, such as glioma treatment plan optimization, has yielded a 27% improvement in two-year patient survival.
The integration of multi-omics technology has led to a significant expansion of application scenarios. Spatial multi-omics reveals the synergistic regulation of methylation and gene expression in the tumor microenvironment, while the digital health platform integrates electronic medical records, wearable devices, and methylation profiles to construct environmental exposure risk models. In the future, breakthroughs in data standardization and privacy protection technologies will accelerate the closed-loop transformation of methylation analysis from laboratory to bedside.
References
- Li, Shizhao, and Trygve O Tollefsbol. "DNA methylation methods: Global DNA methylation and methylomic analyses." Methods (San Diego, Calif.) vol. 187 (2021): 28-43. doi:10.1016/j.ymeth.2020.10.002
- Widschwendter, Martin et al. "Epigenome-based cancer risk prediction: rationale, opportunities and challenges." Nature reviews. Clinical oncology vol. 15,5 (2018): 292-309. doi:10.1038/nrclinonc.2018.30
- Wilhelm-Benartzi, C S et al. "Review of processing and analysis methods for DNA methylation array data." British journal of cancer vol. 109,6 (2013): 1394-402. doi:10.1038/bjc.2013.496




