DNA Methylation Array Principles: Core Mechanisms and Technological Insights
DNA methylation, a "molecular switch" of epigenetic regulation, has been shown to play a role in the control of gene silencing, cell differentiation, and disease processes by dynamically modifying CpG sites in the genome. Conventional testing methodologies, such as sulfite sequencing, can attain single-base precision; however, their clinical application is constrained by cost considerations and the complexity of data processing. The advent of DNA methylation microarray technology, with its high throughput, standardized operation, and cost controllability, has emerged as a pivotal instrument for mapping epigenetic profiles. However, the core challenges of this technology lie in balancing the efficiency of chemical transformation with DNA integrity, optimizing probe hybridization specificity, and eliminating the interference of batch effects on data reliability. In the context of rapid advancements in single-cell epigenomics and liquid biopsy, methylation microarrays are undergoing a pivotal transition from research to clinical translation. This transition entails achieving ultra-high sensitivity in low-abundance samples, such as circulating tumor DNA, and establishing a cross-platform comparable data calibration system. In this paper, we undertake a comprehensive analysis of the molecular mechanisms and technological advancements underlying DNA methylation microarrays. We further elucidate their potential to advance precision medicine from the "gene sequence" to the "epigenetic regulatory layer" through multidisciplinary cross-innovation.
Service you may intersted in
Learn More:
- DNA Methylation Arrays: Principles, Technologies, and Applications
- DNA Methylation Array Development: Evolution, Innovation, and Future Trends
- DNA Methylation Array Workflow & Analysis: Process Optimization and Data Insights
- DNA Methylation Array Applications: Bridging Clinical Breakthroughs and Research Innovations
Fundamental Concepts
DNA methylation, a fundamental mechanism of epigenetic regulation, exerts a profound influence on gene expression and cell fate by dynamically modifying specific regions of the genome. The advent of high-throughput detection technology has rendered DNA methylation arrays a pivotal instrument for the analysis of epigenetic maps. This chapter will provide a comprehensive overview of the biochemical basis, detection principle, and probe hybridization mechanism of DNA methylation arrays. In addition, it will discuss the current optimization strategies and future development directions for this technology. By doing so, it will provide a comprehensive perspective on the scientific core of this technology.
Biochemical Basis of DNA Methylation and Innovations in Detection Techniques
DNA methylation is essentially a covalent modification of the fifth carbon atom (C5) of cytosine, catalyzed by DNA methyltransferases (DNMTs). Within the DNMT family, DNMT1 is responsible for the maintenance of the methylation pattern after replication, whereas DNMT3A/3B establishes the de novo methylation markers during embryonic development. This process relies on the cofactor S-adenosylmethionine (SAM) to provide the methyl group to form 5-methylcytosine (5mC). Methylation regulates gene expression through two pathways: first, it directly hinders the binding of transcription factors (e.g., Sp1) to DNA; second, it recruits methyl-binding proteins (e.g., MeCP2), which, in concert with histone deacetylases (HDACs), compact the chromatin structure and achieve gene silencing.
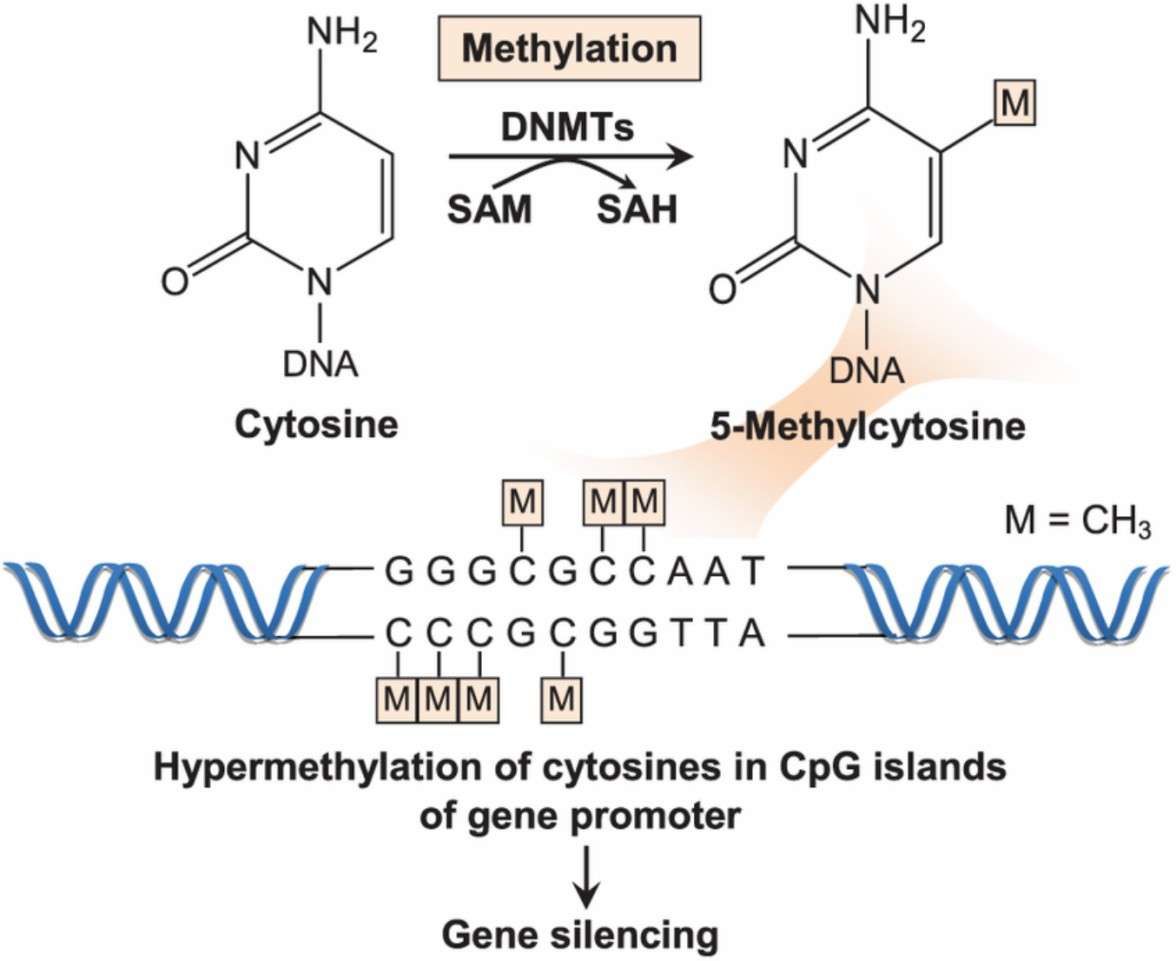 DNMTs catalyse the transfer of a methyl group from the methyl donor SAM to the fifth carbon of the cytosine base in the CpG islands of gene promoter (Wong et al., 2019)
DNMTs catalyse the transfer of a methyl group from the methyl donor SAM to the fifth carbon of the cytosine base in the CpG islands of gene promoter (Wong et al., 2019)
The central challenge in detecting methylation is distinguishing between methylated and non-methylated cytosines. The prevailing technique in the field is based on the conversion of unmethylated cytosine (C) to uracil (U) through the action of sulfite. In contrast, methylated C remains unaltered during this process. Subsequent to amplification by PCR, U is transformed into thymine (T), resulting in discernible signal differences in sequencing or hybridization. In recent years, enzymatic transformation techniques, such as the EZ Methylation Kit developed by Zymo Research, have emerged as an alternative to chemical transformation. These enzymatic techniques reduce DNA breaks by employing mild reaction conditions.
Contemporary technological developments prioritize enhancing resolution and compatibility. For instance, Illumina's methylation microarrays have the capacity to detect nearly a million CpG sites through the design of multiple probes, rendering them suitable for large-scale clinical screening. Conversely, sulfite sequencing has been demonstrated to achieve single-base precision; however, its cost and complexity of data analysis limit its widespread use. The selection of methylation detection technology necessitates a trade-off between resolution, throughput, and cost, with array technology emerging as the platform of choice for epigenetic research due to its exceptional efficiency.
Technological breakthroughs in probe hybridization and signal amplification
The efficacy of methylation arrays is contingent upon the design of probes and the optimization of hybridization procedures. For instance, the Illumina Infinium platform utilizes a two-color probe system to differentiate methylation status. In this system, type I probes are designed with two pairs of probes (matching methylated and unmethylated sequences, respectively) for a specific CpG site, whereas type II probes achieve high-density detection by combining different fluorescent tags (Cy3/Cy5) with a single probe. Probe design necessitates stringent regulation of GC content (40-60%) to circumvent interference from secondary structures and the introduction of concatenated bases (e.g., inosine) to ensure compatibility with sequence variation.
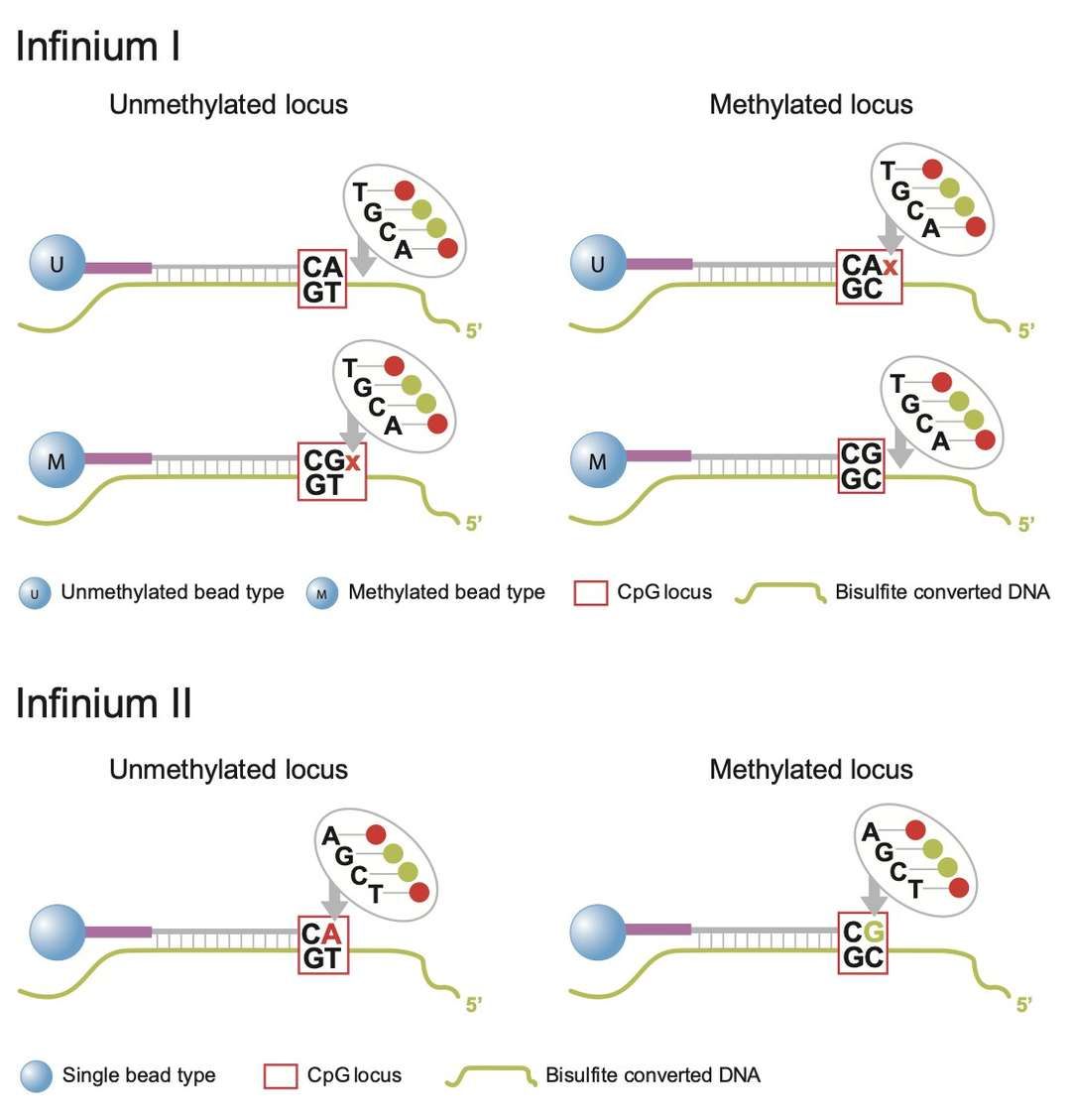 Infinium I and II design
Infinium I and II design
The quantification of signals is contingent upon the calculation of β-value (β = methylated signal / total signal + correction factor), a model that effectively corrects for extreme methylation value bias. To enhance sensitivity, modern arrays integrate enzymatic cascade amplification techniques: Rolled Circle Amplification (RCA) generates repetitive sequences from a circular template and combines them with fluorescent probes to achieve exponential signal growth, while isothermal amplification techniques (e.g., EXPAR) amplify the signal a million-fold in less than 30 minutes. Furthermore, nanomaterials (e.g., gold nanoparticles) leverage the surface plasmon resonance effect to enhance fluorescence intensity or function as probe carriers to improve hybridization efficiency, achieving detection limits as low as 0.05 U/mL.
Optimization of hybridization conditions is also critical. Temperature gradients (e.g., 67°C combined with 15% formamide) have been shown to enhance the specificity of probes with high GC content. Additionally, wash stringency (by adjusting salt concentration) is necessary to optimize the balance between non-specific binding and signal retention. Recent studies have demonstrated that the utilization of tetrahedral DNA scaffolds to regulate probe spacing leads to a reduction in spatial site resistance and enhances signal intensity by a factor of 5-10. The synergistic innovation of probe design and signal amplification technology is driving methylation detection towards higher sensitivity and clinical utility.
Technical Mechanisms
DNA methylation microarray technology, an instrumental tool in the realm of epigenetic research, is predicated on the precise capture of the methylation status of specific CpG sites in the genome through meticulous chemical reactions and probe systems. In this chapter, we will undertake a comprehensive examination of the technical mechanism of this technology, with a particular focus on the chemical principle of disulfide conversion during chip preparation, the technical strategy of methylation detection and quantification, and the cutting-edge exploration directions in this field in the context of current technological development trends.
Chemical cornerstones of chip preparation and disulfide conversion
The preparation of DNA methylation microarrays commences with the extraction and purification of sample DNA, the quality of which directly affects the reliability of subsequent analysis. Following the procurement of high-integrity DNA, disulfide conversion emerges as a pivotal step in distinguishing methylated from unmethylated cytosine. This process entails the selective modification of unmethylated cytosine by sodium disulfide (NaHSO3) under acidic conditions, resulting in its conversion to uracil, while methylated cytosine (5mC) is retained due to the protective effect of the methyl group. The efficiency and specificity of this chemical reaction are contingent on the reaction temperature, pH, and duration. In recent years, researchers have optimized the buffer system (e.g., by adding antioxidants) and the microfluidic reaction device to increase the conversion efficiency to more than 98%, while concomitantly reducing the DNA degradation rate to less than 5%.
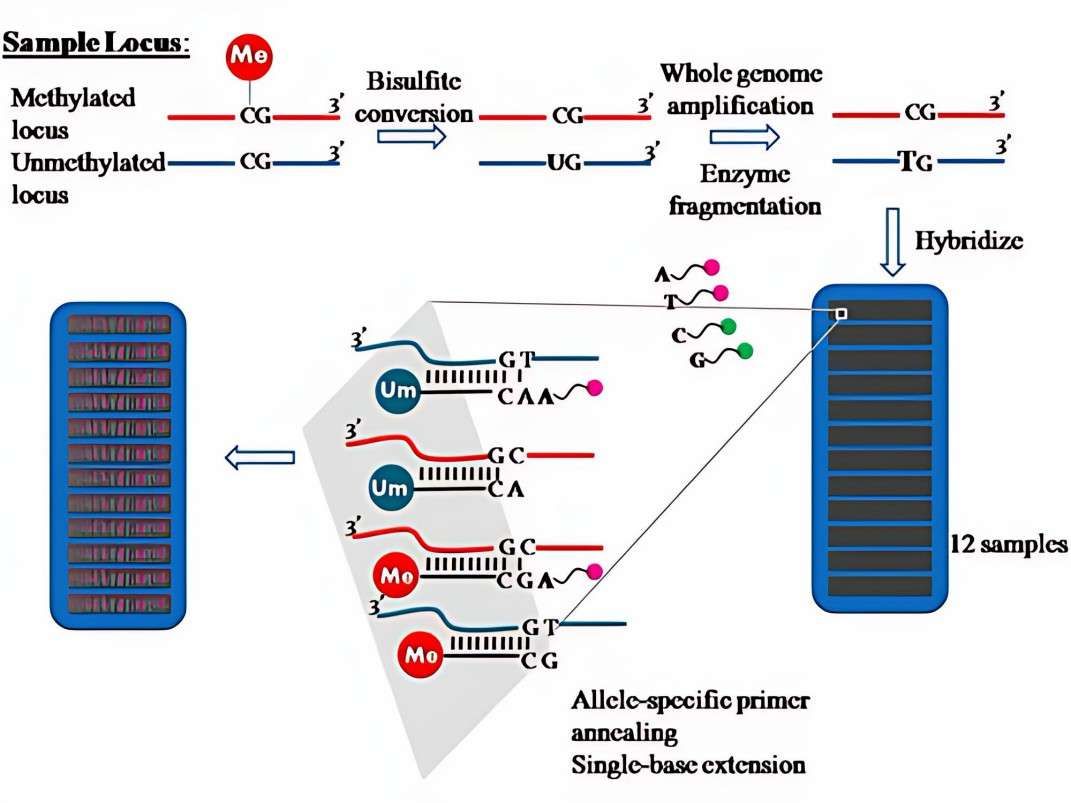 Microarray: DNA Methylation Epic Array
Microarray: DNA Methylation Epic Array
The transformed DNA must undergo amplification via PCR and fragmentation by enzymes to ensure compatibility with the hybridization requirements of the microarray probes. Modern chip designs frequently utilize high-density probe arrays, each meticulously designed to target methylated or unmethylated sequences at a specific CpG site, and labeled with fluorescent dyes for signal capture. For instance, the Illumina Infinium MethylationEPIC chip has the capacity to address over 850,000 CpG sites, and its probe design integrates single-base extension technology to markedly enhance detection throughput and precision. The core value of disulfide conversion is to establish a "molecular label" of methylation status through chemical modification, which lays the foundation for subsequent hybridization and signal resolution.
Technical Strategies for Methylation Detection and Quantification
Following the completion of the microarray hybridization process, the resolution of the methylation level is contingent upon the implementation of diversified detection techniques. Among these methods, the fluorescence signal detection method is considered a mainstream approach. This method directly reflects the methylation level of target sites by scanning the difference in fluorescence intensity of probes on the chip. The Illumina iScan system, for instance, utilizes a dual-color fluorescence channel (Cy3/Cy5) to detect methylated and unmethylated alleles simultaneously. This is then combined with normalization algorithms (e.g., β-value calculation) to achieve quantitative analysis. In recent years, the advent of single-molecule fluorescence imaging technology has enabled detection sensitivity up to single-copy resolution, a development that is particularly well-suited for epigenetic analysis of low-abundance samples, such as circulating tumor DNA.
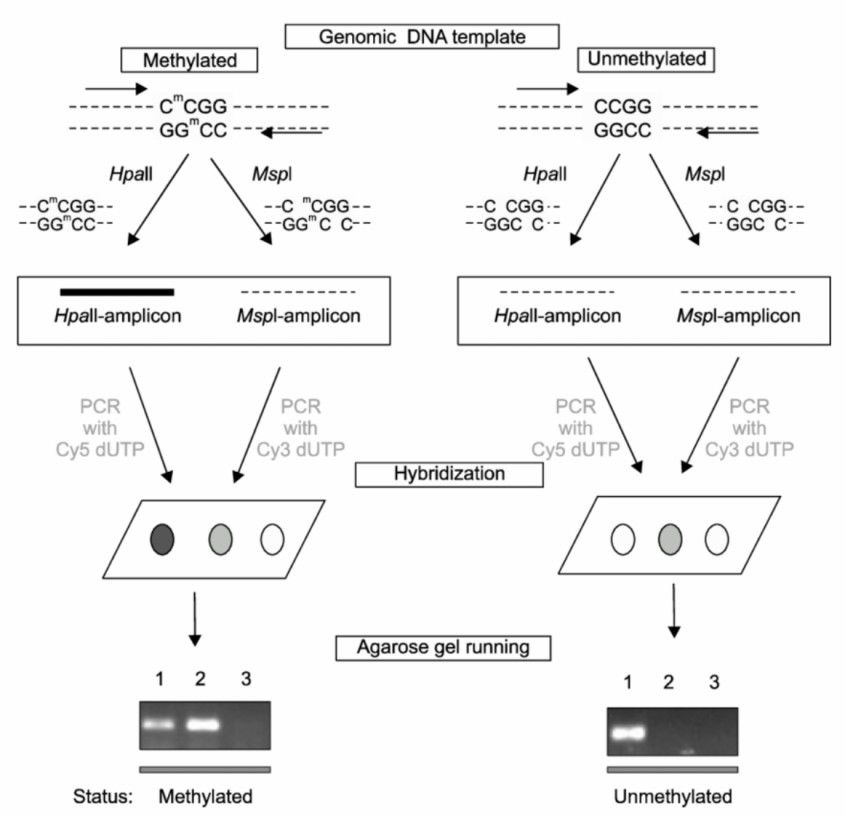 Schematic diagram of methylation microarray (Kwon et al., 2008)
Schematic diagram of methylation microarray (Kwon et al., 2008)
The integration of high-throughput sequencing technologies has further expanded the boundaries of microarray applications. Targeted sequencing methods, such as TBS-seq, have emerged as a significant advancement, facilitating deep sequencing by capturing the region of the microarray probe. This approach enables the verification of the methylation status at the single-base resolution, thereby circumventing the constraints imposed by microarray probe coverage limitations. Furthermore, quantitative PCR (qPCR) maintains its relevance in small-scale validation and clinical diagnostics due to its high sensitivity and cost-effectiveness. For instance, methylation-specific PCR (MSP) can achieve detection at 1% methylation allele frequency by designing methylation-dependent primers, thereby providing technical support for early cancer screening. Current technological trends are moving toward multi-omics integration, such as combining chromatin accessibility data (ATAC-seq) with methylation microarray results to construct a three-dimensional epigenetic regulatory network, thus more comprehensively resolving gene silencing mechanisms.
Optimizing Core Principles
The precision of DNA methylation microarray technology is contingent upon the meticulous optimization of the experimental process and the rigorous calibration of data analysis. As epigenetic research progresses towards single-cell resolution and clinical translation, the necessity of enhancing hybridization specificity, mitigating background interference, and establishing a more robust data standardization system through technological advancement has become a pivotal concern in this domain. This chapter will systematically review the latest advances in hybridization optimization strategies and data calibration techniques, and reveal how they can jointly facilitate the transition from "qualitative" to "quantitative" methylation detection.
Optimization of hybridization conditions
The enhancement of hybridization efficiency and signal-to-noise ratio of microarrays is contingent upon the molecular engineering optimization of probe design. Modern methylation microarrays commonly use probes of 50-70 base pairs (bp) in length, whose sequences are rigorously screened to avoid cross-reactivity with non-target regions. For instance, the Illumina MethylationEPIC v2 chip employs "probe shielding" technology, which involves the prediction of potential non-specific binding sites through the calculation of thermodynamic parameters (e.g., ΔG values) and the subsequent insertion of blocking sequences to suppress background signals. In regard to the kinetic control of hybridization reactions, researchers have reduced the melting temperature (Tm) of double-stranded DNA by adjusting the formamide concentration (typically 25%-40%) to make the binding of probes to target sequences more selective. Furthermore, the optimized ratios of sequestering agents (e.g., Cot-1 DNA and salmon sperm DNA) effectively adsorb repetitive sequences in the samples, thereby reducing nonspecific hybridization background by more than 30%.
In recent years, the application of microfluidic chip technology has provided a novel approach for the dynamic regulation of hybridization conditions. The integration of a temperature gradient module and a real-time fluorescence monitoring system enables the synchronous testing of multiple sets of hybridization conditions (e.g., salt ion concentration, pH value) on a single chip. This approach facilitates the rapid screening of optimal parameter combinations. This "Lab-on-a-chip" strategy not only reduces the method development cycle, but also enhances batch-to-batch consistency, making it particularly well-suited for large-scale clinical sample testing.
Data standardization and calibration
The accuracy of methylation microarray data is contingent upon the robustness of the normalization algorithm. Conventional β-value (β=methylated signal/(methylated signal + unmethylated signal + α)) calculations are vulnerable to fluctuations in fluorescence intensity. However, novel normalization methods, such as SWAN (Subset-quantile Within Array Normalization), have been developed to address these limitations. SWAN utilizes quartiles to correct for technical variations between probe types (Infinium I/II), thereby reducing the impact of batch effects by more than 50%. In the context of single-cell methylation microarray data, researchers have developed imputation algorithms based on deep generative models (e.g., VAE). These imputation algorithms effectively fill in missing values due to insufficient DNA amount while preserving cellular heterogeneity characteristics.
The establishment of the calibration system is contingent upon the enhancement of standard reference materials (SRMs). The Methylation Reference Material (RM 8375), developed by the National Institute of Standards and Technology (NIST), comprises 12 DNA fragments with known methylation ratios, spanning a methylation gradient from 0% to 100%, thereby serving as a "ruler" for inter-laboratory data comparison. In clinical applications, the methylation quantification of liquid biopsy samples is often affected by plasma free DNA fragmentation. To address this challenge, a dynamic calibration model based on fragment length correction factor (FLCF) has been proposed. This model aims to enhance the precision of methylation level estimation in plasma samples by integrating data on the size distribution of DNA fragments, thereby reducing the estimation error from ± 8% to ± 3%. Current technological trends are moving toward multimodal calibration, such as combining methylation microarrays with nanopore sequencing data to construct joint calibration curves, thus breaking through the resolution limitations of a single technology platform.
Conclusion
From the micro-regulation of hybridization kinetics to the macro-calibration of the data pipeline, the optimization of methylation microarray technology has always been centered on the two core elements of "precision" and "robustness." The advent of AI-driven adaptive hybridization systems (e.g., reinforcement learning-based temperature control models) and organ-specific methylation reference databases will not only enable higher throughput in future methylation assays but will also facilitate the maintenance of a balance between sulfite conversion efficiency and hybridization specificity in complex biological matrices (e.g., FFPE tissues). These technological breakthroughs will accelerate the translation of methylation biomarkers from laboratory discovery to clinical diagnostic applications, providing a more reliable epigenetic basis for early cancer screening and personalized treatment.
References
- Wong, Kah Keng et al. "Oncogenic Roles and Inhibitors of DNMT1, DNMT3A, and DNMT3B in Acute Myeloid Leukaemia." Biomarker insights vol. 14 1177271919846454. 8 May. 2019, doi:10.1177/1177271919846454
- Kwon, Mi et al. "Identification of DNA Methylation Markers for NSCLC Using Hpall-Mspl Methylation Microarray." Tuberculosis and Respiratory Diseases. 65(2008). doi:10.4046/trd.2008.65.6.495




