
Core Advantages of Direct RNA Methylation Sequencing
By sequencing native RNA directly as it passes through the nanopore, this technology circumvents the artificial artifacts introduced by standard library preparation kits, providing unparalleled insights into the epitranscriptome.
- One-Shot Multi-Omics Data: Maximize the value of your precious samples. A single sequencing run simultaneously captures gene expression levels, full-length alternative splicing structures, dynamic poly(A) tail lengths, and diverse base modifications (such as m6A and m5C).
- Zero Amplification Bias: By completely eliminating reverse transcription and PCR amplification, Direct RNA Sequencing provides an absolute, unbiased representation of RNA abundance. This is particularly crucial for resolving transcripts with extreme GC content or highly complex secondary structures that typically fail in short-read workflows.
- High-Accuracy Basecalling and Decoding: Harnessing advanced neural network algorithms, our pipeline accurately translates raw electrical current fluctuations into base-specific modification maps, drastically reducing the false-positive rates traditionally associated with early nanopore technologies.
Spotlight on RNA Methylation: Decoding the Epitranscriptome at Single-Base Resolution
While capturing full-length transcript structures is highly valuable, the true transformative power of Direct RNA Sequencing lies in its ability to unmask the hidden epigenetic layer of RNA. RNA modifications, collectively known as the epitranscriptome, play indispensable roles in regulating mRNA stability, nuclear export, translation efficiency, and alternative splicing.
Historically, profiling critical modifications like N6-methyladenosine (m6A) or 5-methylcytosine (m5C) required immunoprecipitation techniques. These methods demand massive RNA input, suffer from antibody cross-reactivity, and only localize modifications to rough "peak" regions (100–200 bases wide) on heavily fragmented RNA.
Our Direct RNA Sequencing service fundamentally resolves these limitations, offering a dedicated, high-resolution view of RNA methylation:
- Single-Molecule, Base-Specific Precision: Because modifications physically alter the electrical current as the native RNA molecule translocates through the nanopore, we can identify the exact nucleotide harboring the modification. This base-specific resolution allows for the discovery of precise regulatory motifs that regional enrichment techniques miss.
- Simultaneous Multi-Modification Detection: Unlike IP-based methods that require a separate sequencing run and a specific antibody for every single modification type, DRS captures the raw electrical signature of the entire molecule. Advanced algorithms can theoretically decode multiple distinct modification types (e.g., m6A, m5C, pseudouridine) from a single dataset, providing a holistic view of the epitranscriptomic landscape.
- Phasing Modifications to Specific Isoforms: This is the ultimate analytical breakthrough. Short-read methods cannot tell you which splice variant carries a specific m6A mark. By reading the molecule from end-to-end, our service definitively links specific methylation events to distinct, full-length alternative splicing isoforms. This allows researchers to definitively prove, for example, whether methylation at a specific locus directly triggers intron retention or exon skipping.
Applications of Direct RNA Methylation Sequencing
The ability to directly read native RNA molecules opens up novel avenues across diverse biological disciplines. By preserving both structural integrity and epigenetic markers, this service accelerates discoveries in areas where traditional short-read sequencing falls short. Understanding the spatial and temporal distribution of RNA modifications is no longer a niche pursuit; it is central to decoding cellular function across the broader epigenetics research spectrum.
The dysregulation of RNA methylation machinery (writers, erasers, and readers) is a hallmark of many cancers. Map m6A, m5C, and other critical RNA modifications at single-nucleotide resolution to uncover post-transcriptional regulatory networks that drive tumorigenesis, metastasis, and drug resistance. Direct RNA sequencing allows researchers to pinpoint which specific transcript variants carry aberrant modifications.
The mammalian brain expresses the most complex transcriptome, characterized by extensive alternative splicing. Accurately resolve these highly complex alternative splicing events and identify novel fusion genes without the risk of PCR chimeras or assembly errors. Linking these splice variants to specific epitranscriptomic marks helps elucidate mechanisms of neuronal plasticity and neurodegeneration.
Plants rely heavily on rapid transcriptional reprogramming to survive environmental changes. Investigate how environmental stressors (drought, heat, salinity) or developmental cues dynamically alter RNA methylation and isoform usage in crop species and model organisms. This dual-layer mapping is essential for engineering stress-resilient agricultural varieties.
RNA viruses utilize sophisticated epitranscriptomic strategies to hijack host machinery and evade immune detection. Directly sequence viral RNA genomes to identify functional modification sites that mediate viral replication and host immune evasion, bypassing the challenges of overlapping open reading frames and high mutation rates that confound short-read assembly.
Non-coding RNAs are master regulators of chromatin dynamics and gene expression. Characterize full-length long non-coding RNAs (lncRNAs) and map their specific epigenetic modifications to understand their roles in gene regulation. Direct sequencing is particularly vital here, as many lncRNAs lack poly(A) tails and form complex secondary structures that resist standard reverse transcription.
Direct RNA Sequencing Workflow & Strict QC Checkpoints
A successful Direct RNA Sequencing project requires immaculate handling of highly sensitive native RNA. Our laboratory executes a rigorous, fully optimized workflow designed to preserve RNA integrity and maximize data output.
- Strict RNA Extraction & Initial QC: Upon sample receipt, we perform rigorous quality control using Agilent Bioanalyzer/TapeStation systems to confirm RNA Integrity Number (RIN) and exact concentration.
- Poly(A)+ Enrichment: To maximize sequencing efficiency for protein-coding genes, total RNA undergoes specialized Oligo d(T) bead enrichment to isolate intact polyadenylated messenger RNA.
- PCR-Free Library Preparation: Specialized motor proteins and sequencing adapters are directly ligated to the native 3' and 5' ends of the RNA molecules. Absolutely no reverse transcription or PCR amplification occurs.
- Nanopore Sequencing: The adapted RNA libraries are loaded onto specialized flow cells. As the motor protein feeds the native RNA strand through the nanopore, disruptions in the ionic current are recorded in real-time.
- Signal Decoding & Data Delivery: Raw electrical signals are securely transferred to our computing clusters, where neural networks decode the current into base sequences and modification probabilities.

Comprehensive Bioinformatics Analysis: From Basecalling to m6A-Isoform Networks
The true challenge of Nanopore Direct RNA Sequencing does not lie solely in the wet lab, but in the immense computational power required to decode raw Fast5/Pod5 electrical signals into biologically meaningful data. We provide an end-to-end bioinformatics pipeline tailored to dissect complex RNA methylation networks.
We believe in complete data transparency. You will receive all raw signal files, allowing your internal biostatistics teams to train custom machine-learning models in the future if desired. Based on our rigorously validated algorithms, we deliver a highly structured analysis package spanning four core analytical modules. These modules are designed to transition raw signal data into publication-ready biological insights.
I. Isoform Analysis
Understanding the precise architecture of the transcriptome is the first step in epitranscriptomic analysis. We utilize long-read alignment tools to map reads to the reference genome, minimizing the ambiguity of short-read assembly.
- Alternative Splicing Analysis: Identification and categorization of complex splicing events (exon skipping, intron retention, alternative 5'/3' splice sites) at the full-length transcript level. This prevents the generation of chimeric artifacts common in PCR-based methods.
- Fusion Gene Identification: High-confidence detection of fusion transcripts. Because single reads span the entire transcript, we can confidently identify fusion breakpoints without relying on complex, error-prone assembly algorithms.
- SSR Analysis: Simple Sequence Repeat (SSR) mapping within transcript sequences, providing valuable markers for genetic diversity and mapping studies.
- LncRNA Analysis: Identification and structural characterization of long non-coding RNAs, differentiating them from protein-coding transcripts based on coding potential calculations.
- Poly(A) Analysis: Precise estimation and tracking of dynamic poly(A) tail lengths directly from the electrical signal duration. This provides critical data for assessing mRNA stability, decay rates, and translational efficiency without requiring separate, labor-intensive assays.
II. Expression Quantification
Accurate quantification relies on the zero-amplification nature of the DRS workflow, ensuring that read counts directly correlate with physical RNA molecule abundance.
- Transcript Quantification: Unbiased abundance calculation for full-length transcripts. We report normalized expression values (e.g., TPM or CPM) suitable for cross-sample comparison.
- Differential Transcript Analysis: Statistical evaluation of transcript expression changes across varying conditions using robust models (e.g., DESeq2 applied to long-read counts) to identify statistically significant shifts in the transcriptome.
- Enrichment Analysis: Functional mapping of differentially expressed transcripts to Gene Ontology (GO) terms and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways, contextualizing the transcriptomic changes within broader biological functions.
- Protein-Protein Interaction (PPI) Networks: Predicting interactive networks based on the translated products of the quantified transcriptomes, helping to identify key hub genes and regulatory modules.
III. Methylation Modification Analysis
This module represents the core advantage of Direct RNA Sequencing. We utilize specialized neural networks trained on vast datasets of modified and unmodified synthetic sequences to calculate modification probabilities at each base.
- m6A Site Annotation: Base-resolution mapping of modification sites. We annotate these sites across different structural regions of the RNA (e.g., 5' UTR, CDS, 3' UTR) to help deduce their potential regulatory function.
- Methylation Modification Enrichment Analysis: Evaluating the functional enrichment of genes harboring specific modification sites. Are methylated genes disproportionately involved in specific pathways, such as stress response or cell cycle regulation?
- Differential m6A Analysis: Rigorous statistical evaluation of how methylation landscapes shift in response to disease states, environmental stress, or gene knockouts. We identify sites that are significantly hyper- or hypo-methylated between your experimental groups.
IV. Isoform Joint Quantitative Expression Analysis
This advanced module synthesizes structural and epigenetic data, allowing researchers to answer complex questions about how modifications influence transcript fate.
- AltTP Analysis: Evaluation of Alternative Transcript Proportion. This determines whether the dominant isoform of a gene shifts under different conditions, providing insights into splicing regulation.
- Functional Diversity Analysis (FDA): Correlating structural diversity with specific epitranscriptomic markers. We analyze whether transcripts with higher diversity in splicing also exhibit distinct methylation patterns.
- Differential Enrichment Analysis: Multi-dimensional integration linking epigenetic changes directly to altered transcript fate. For example, we can determine if a specific m6A site consistently correlates with the retention of a specific intron across multiple samples.
Publication-Ready Demo Results
Our deliverables are designed not just for raw data storage, but to be directly integrated into your upcoming manuscripts. Our analysis packages generate highly visual, statistically rigorous representations of your epitranscriptomic data.
- Isoform Differential Heatmaps: Intuitively visualizes the significant differential expression of full-length transcripts across varied conditions, providing a macro-view of transcriptomic shifts.
- m6A Modification Site Distribution (Metagene Plots): Precise, single-nucleotide resolution mapping of m6A sites plotted along the standardized RNA structure, immediately highlighting whether modifications are enriched in UTRs or coding sequences.
- Isoform & Methylation Joint Analysis Networks: Integrative network plots showcasing Alternative Transcript Proportion (AltTP) and functional diversity, visually correlating specific methylation events with dominant splice variants.
- Poly(A) Tail Length Distribution (Violin Plots): Tracks the dynamic fluctuation of poly(A) tail lengths, offering critical visual evidence for studies focusing on mRNA stability, degradation pathways, and translation efficiency.
- Full-Length Transcript Differential Expression: Classic, high-contrast volcano plots that highlight significantly altered genes at the full-transcript level, ensuring rapid identification of top candidates.
Sample Requirements
Because Direct RNA Sequencing analyzes the native molecule without any amplification steps to boost low-input signals, the starting material requirements are significantly higher than standard short-read sequencing. Ensuring your samples meet these criteria is the most critical step toward project success.
| Sample Type | Recommended Input | Concentration | Purity (OD260/280) | Notes |
|---|---|---|---|---|
| Total RNA | > 20 μg | ≥ 180 ng/μL | 1.8 - 2.2 | Ensure high integrity (RIN > 7.0 strictly recommended). Avoid multiple freeze-thaw cycles. |
| Cell Suspension | ≥ 1x10^7 cells | N/A | N/A | Must be flash-frozen immediately or stored in an appropriate validated RNA stabilizer. |
| Tissue (Animal/Plant) | > 100 mg | N/A | N/A | Flash-freeze in liquid nitrogen immediately upon harvest. |
If you are working with highly degraded samples or find it difficult to reach the 20 μg threshold, please consult our upstream RNA extraction services for specialized low-input protocol support before abandoning your project.
Technology Decision Guide: DRS vs. MeRIP-Seq vs. Iso-Seq
Why Choose DRS for RNA Methylation Analysis
Selecting the right platform is critical for any RNA methylation project. For studies that require native RNA context, full-length transcript information, and amplification-free analysis, Nanopore Direct RNA Sequencing (DRS) is often the preferred strategy. The comparison below is designed to help researchers determine when DRS is the strongest fit, and when alternative approaches such as MeRIP-Seq or PacBio Iso-Seq may be more appropriate.
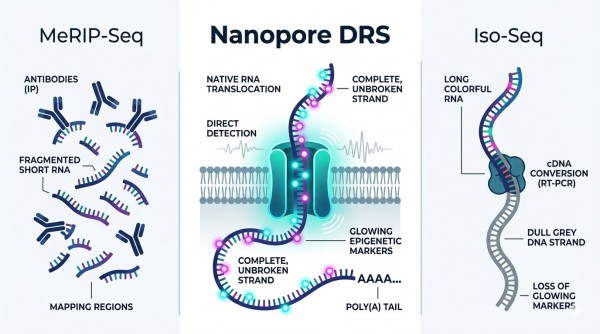
Comparison of Transcriptomic Methodologies
| Target Dimension | Nanopore DRS | MeRIP-Seq (Antibody IP) | PacBio Iso-Seq |
|---|---|---|---|
| Best For | Native RNA methylation analysis with isoform context | Cost-effective profiling of known modifications across larger cohorts | High-accuracy full-length transcript structure analysis |
| Modification Readout | Direct, native RNA signal with modification-aware analysis | Antibody-enriched signal for known marks (e.g., m6A) | No native modification information |
| Isoform Context | Yes | No | Yes |
| Resolution | Single-nucleotide / single-molecule potential | Peak region (typically 100-200 bp) | Base-level sequence only |
| Amplification Bias | Minimal, no PCR amplification | Higher, due to enrichment and amplification steps | Present, due to cDNA synthesis and amplification |
| Retains Native RNA Features | Yes | No | No |
| Poly(A) Tail Information | Yes | No | No |
How to Choose the Right Platform
Choosing the most suitable method depends on your study objective, sample type, and desired level of biological resolution.
- Choose Nanopore DRS when your project requires native RNA analysis, transcript isoform resolution, and the ability to investigate candidate RNA methylation events in their full-length transcript context. DRS is particularly well suited for studies exploring how RNA modifications may be associated with alternative splicing, transcript stability, or poly(A) tail dynamics.
- Choose MeRIP-Seq when your main goal is cost-effective, region-level profiling of a known modification across a larger sample cohort, and full-length transcript context is not required. This is often a practical option for broad enrichment-based screening.
- Choose PacBio Iso-Seq when you need highly accurate full-length transcript structures and splice junction discovery, but native RNA modification analysis is not part of your primary objective.
If Nanopore DRS is not the best fit for your project goals, throughput needs, or sample characteristics, our team can also help guide you toward alternative workflows such as antibody-based enrichment or cDNA long-read sequencing.
Case Study: Decoding Plant Epitranscriptomics with Direct RNA Sequencing
Frequently Asked Questions (FAQ)
Q: Why is >20μg of Total RNA strictly required for Direct RNA Sequencing?
A: Unlike standard RNA-Seq, which relies on heavy PCR amplification to generate millions of library copies from nanograms of input, Direct RNA Sequencing measures the exact, physical RNA molecules present in your tube. Because only a fraction of Total RNA is Poly(A)+ mRNA (typically 1-5%), starting with 20μg ensures we can capture enough native mRNA molecules to achieve the necessary sequencing depth for meaningful statistical analysis.
Q: How does your bioinformatics pipeline distinguish m6A from other RNA modifications?
A: Distinct RNA modifications (e.g., m6A, m5C, pseudouridine) physically alter the ionic current in unique, measurable ways as they pass through the nanopore. We utilize advanced, continually updated deep-learning models trained specifically on vast datasets of known synthetic modifications. These algorithms analyze the shape, dwell time, and intensity of the current variations to calculate a high-confidence probability score for specific modification types at single-base resolution.
Q: Will I receive the raw Fast5/Pod5 files containing the electrical current signals?
A: Yes. We believe researchers should retain complete ownership and flexibility over their data. In addition to the processed FastQ files and final bioinformatics reports, we provide the raw Fast5/Pod5 electrical signal files. This ensures your bioinformatics team can re-analyze the data in the future as basecalling algorithms inevitably improve or if you wish to train custom models for entirely novel epigenetic markers.
References
- Direct RNA sequencing enables m6A detection in endogenous transcript isoforms at base-specific resolution
- Nanopore direct RNA sequencing maps the complexity of Arabidopsis mRNA processing and m6A modification
- Native RNA or cDNA Sequencing for Transcriptomic Analysis: A Case Study on Saccharomyces cerevisiae
- Identification of the flowering repressor FIONA1 by whole-genome re-sequencing
Disclaimer: The services and products described on this page are for Research Use Only (RUO) and are not intended for use in diagnostic or therapeutic procedures.