
Common Research Thoughts of De novo in Animal and Plant Genome
De novo sequencing of animals and plants, means that a species can be sequenced without any reference sequence information, and the genome sequence map of the species can be obtained by splicing and assembling with bioinformatics analysis methods. Using the whole genome de novo sequencing technology, we can obtain the whole genome sequence of animals and plants, which will promote a series of downstream research of this species, thus promoting the research of this species. After the whole genome sequence map is completed, the genome database of this species can be constructed, which will build an efficient platform for post-genomics research of this species and provide DNA sequence information for subsequent gene mining and functional verification.
With the development of sequencing technology, a variety of representative sequencing technologies such as second-generation sequencing (illumina, MGI, etc.), third-generation sequencing (PacBio, Nanopore, etc.), optical mapping, Hi-C mapping and their combination assembly schemes have appeared, which have greatly reduced the difficulty and cost of genome assembly and accelerated the mining of species genome data.
In the field of genomics research, a key and basic link is to choose the correct sequencing strategy. Faced with a wide variety of sequencing technologies and complex research purposes, researchers need to comprehensively consider many factors, such as the characteristics of samples, the nature of research problems, budget constraints and time requirements, so as to accurately choose the sequencing strategy that suits their own research. At the same time, with the deepening of genomics research, it is particularly important to expand the research ideas of genomics. This will not only help to break through the limitations of traditional research and tap more potential biological information, but also provide a new perspective and method for solving many biological problems such as disease pathogenesis and biological evolution. So, how to choose the correct sequencing strategy, and what ways can expand the research ideas of genome?
 de novo common research ideas
de novo common research ideas
Service you may interested in
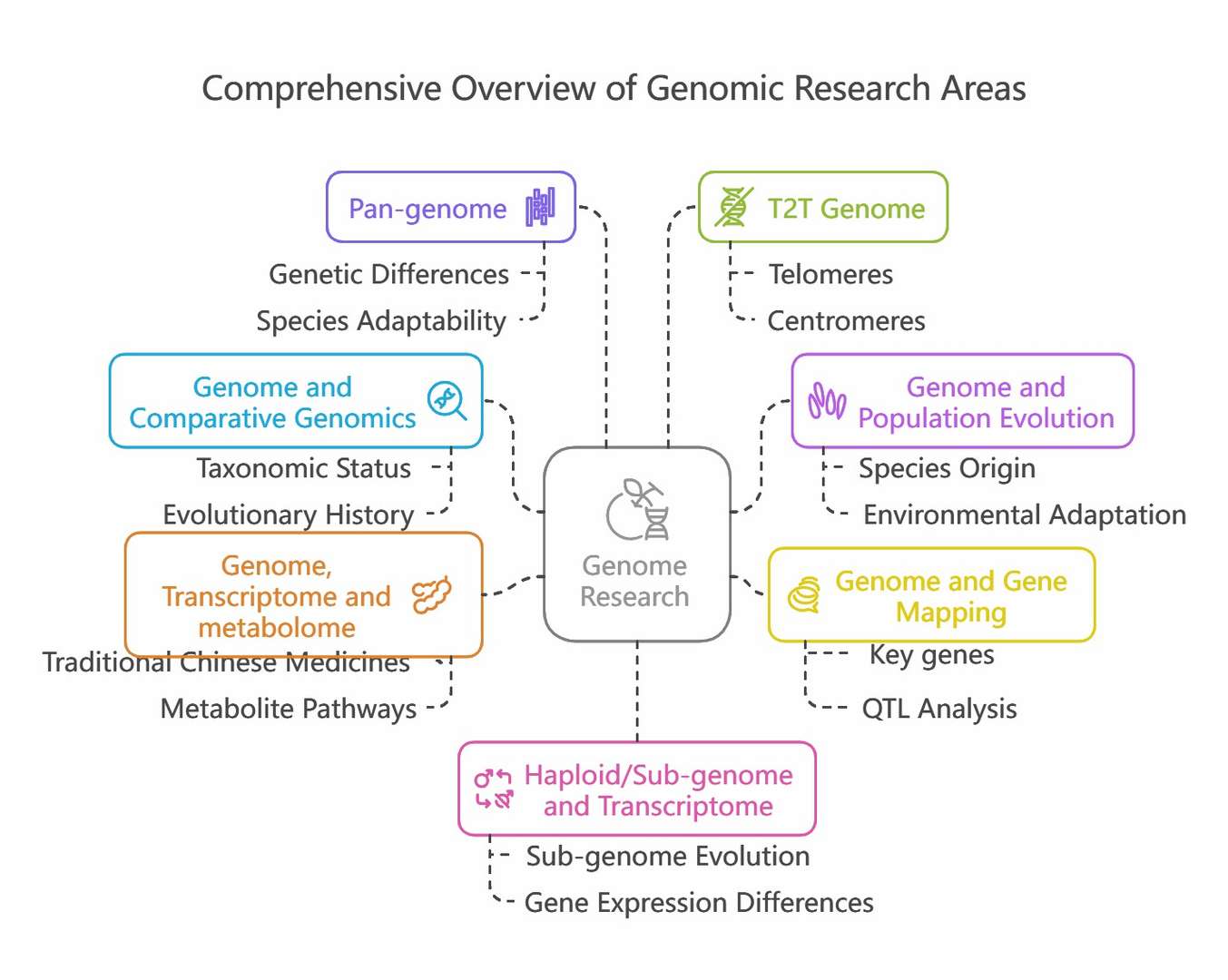
Genome and Comparative Genomics
Crassostrea hongkongensis is a unique and dominant economic aquaculture species along the coast of South China, with a typical fixed lifestyle and a high degree of asymmetry between the left and right shells. Therefore, based on technologies such as illumina, PcaBio and Hi-C, this study finally completed the high-quality genome assembly of 729.6 Mb Hong Kong oyster. Phylogenetic analysis shows that oyster species were formed about 92.1 million years ago, starting at the end of Cretaceous-Paleogene extinction events. Through the joint analysis with comparative genome, it was found that the loss of homeobox gene Antp and the extensive expansion of extracellular matrix gene family were the key to drive oyster from attachment to fixation. Asymmetric expression of homologous transcription factors (Pitx2 and Rfx6) and strain-specific expansion gene family tyrosinase may be the molecular basis of shell asymmetry. The research results provide an important reference for understanding the biological evolution and biological protection of bivalves.
 The genome landscape and phylogenetic analysis of the oyster crassostrea hongkongensis (Zhang et al., 2022)
The genome landscape and phylogenetic analysis of the oyster crassostrea hongkongensis (Zhang et al., 2022)
Genome Combined with Population Evolution
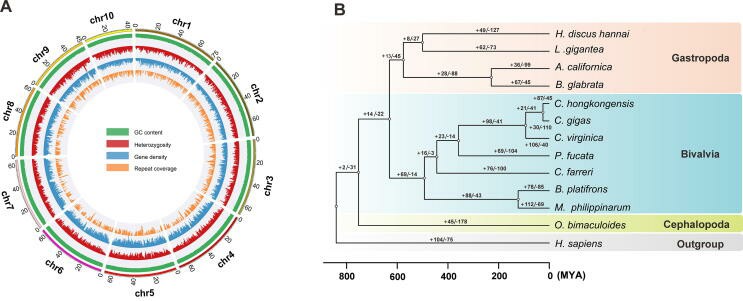
Four apricot germplasm resources (Marouch #14 and Stella from P. Armenia, CH320_5 from Asia and CH264_4 from Siberia) were assembled from scratch by using Pacbio-sequencing, Nanopore sequencing, illumina sequencing and Bio-Nano optical mapping technology, and high-quality reference genomes were obtained. In order to study the genetic diversity and evolution of apricot and its wild relatives, 564 resource materials were re-sequenced, including 256 wild P. armeniaca materials from natural populations in Central Asia, 43 wild P. sibirica materials from 8 natural populations in China, 1 P. mandshurica material and 264 P. armeniaca materials (27 from China, 166 from Europe, 71 from Central Asia).
At the same time, 14 P. brigantina materials were selected as the outer group. The evolutionary history of apricot gene pool was reconstructed by ABC-RF method. The results showed that there was gene flow among different species, which initially differentiated from P. mume population to P. sibirica wild population, and then further differentiated into P. armeniaca wild southern population and P. armeniaca wild northern population. The cultivated species in China came from the wild southern population of P. armeniaca, while the cultivated species in Europe came from the wild northern population of P. armeniaca, so it can be inferred that the cultivated species in China and the cultivated species in Europe evolved independently. Then, the genome selection regions of European and China cultivars were identified by CLR, Fst, πW/πCultivated results. By analyzing the genes in these selected regions, it was found that European cultivars and China cultivars adopted different selection strategies in the domestication process. European cultivars focus on the selection of fruit size and acidity during domestication, while China cultivars focus more on the selection of resistance.
 Geographical distribution and features of Armeniaca species (Groppi et al., 2021)
Geographical distribution and features of Armeniaca species (Groppi et al., 2021)
Genome Together with Gene Mapping
Pacbio-sequencing, 10x Genomics sequencing, illumina sequencing, HiC mapping and Bionano optical mapping were used to assemble diploid Camellia CON(2n=30) materials from scratch, and 2.89Gb Camellia genome was obtained, with contig N50=1.002 Mb, and 91.33% of the sequences were anchored to 15 chromosomes. A total of 42,426 protein coding genes were obtained by annotation, with an average length of 3955 bp and a repetitive sequence content of 69%.
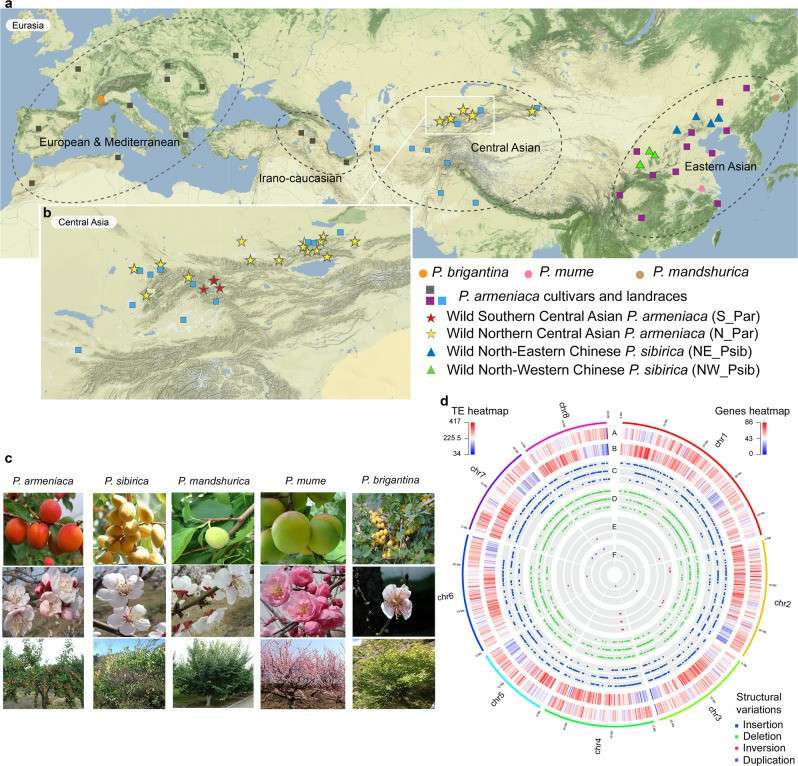
In order to evaluate the domestication characteristics of Camellia oleifera seed oil, the researchers studied a natural population, which contained 221 materials from different regions of China. Phenotypic analysis showed that there were significant differences among different Camellia oleifera varieties, among which oleic acid content was negatively correlated with linoleic acid content. Subsequently, the researchers sequenced the transcriptome of 221 materials, and obtained 1849953 SNPs and 85440 InDel through comparative analysis. The researchers conducted GWAS analysis, quantitative GWAS-qGWAS analysis and eQTL analysis on 221 cultivated Camellia oleifera, among which 711 genes were identified in 100Kb region by GWAS analysis. QGWAS analysis identified 204 genes related to oil traits.
EQTL analysis showed that 9001 transcripts were cis-eQTL and 6,548,567 trans-eQTL, and the analysis of gene co-expression network was consistent with EQTL results. The results of GWAS and qGWAS were analyzed by overlap, and 21 candidate genes with high reliability were identified, 14 of which were related to total oil content. Specifically, 9 of these 14 genes are involved in lipid metabolism, and 5 are plant hormone-related transcription factors. Finally, the transcriptome analysis of the above 14 genes showed that SDP1, IAA26, FabD and Oleosin3 were related to the domestication of Camellia oleifera, SAC8 and KASIII were related to palmitic acid content, and GDL57, GLPK and SADs were related to stearic acid content.
In order to further determine the effective allelic variation of candidate genes, the researchers analyzed the contribution of nucleotide polymorphism of candidate genes to the variation of oil traits, and determined that the candidate genes of eight SNP combinations were significantly related to the changes of oil traits and gene expression. These eight genes have significant single nucleotide polymorphism and expression level differences in cultivated populations, and they include both genes related to oil synthesis and oil decomposition, which shows that the oil traits of Camellia oleifera are the result of the coordination of genes related to oil synthesis and decomposition. This study provides new insights for the domestication and genetic breeding of Camellia oleifera.
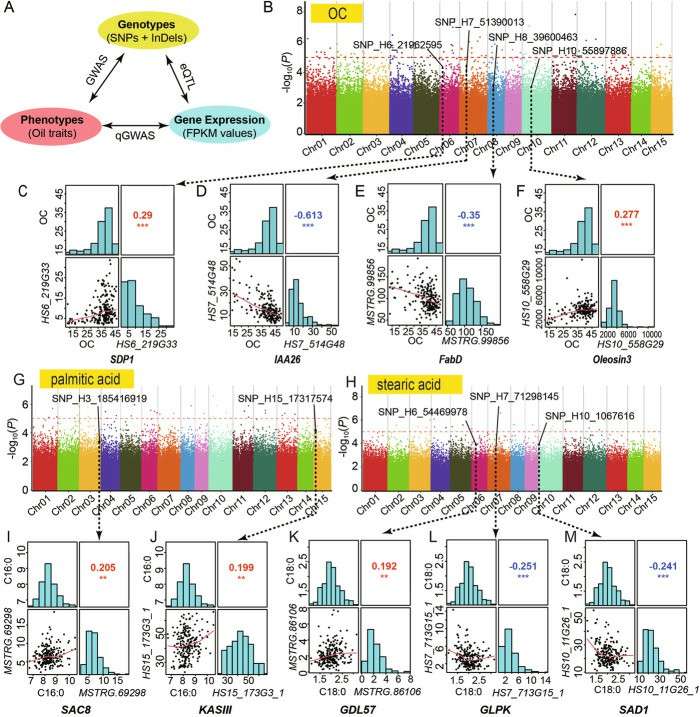 Genome-wide identification and annotations of candidate genes involved in the seed oil domestication (Lin et al., 2022)
Genome-wide identification and annotations of candidate genes involved in the seed oil domestication (Lin et al., 2022)
Genome, Transcriptome and Metabolome
Seabuckthorn (Hippophae) is an important economic and ecological species in Hippophae, which is rich in biological and pharmacological active substances. Through illumina, Pacbio and Hi-C sequencing, the genome assembly of Hippophae rhamnoides was completed, and the first genome sequence of Elaeagnaceae was obtained, with a total length of 849.04MB, an N50 of 69.52MB and an annotation gene of 30864. It is inferred that two chromosome doubling events occurred, one about 36-41 million years ago (Mya) and the last about 24-27 MYA, resulting in gene amplification related to ascorbic acid and aldehyde acid metabolism, lipid biosynthesis and fatty acid extension.
Comparative transcriptomics and metabolomics analysis identified some key genes leading to high content of polyunsaturated fatty acids and ascorbic acid. In addition, through population resequencing of 55 materials, 9.8 million genetic variations were identified in seabuckthorn germplasm. The genes obtained by selective scavenging analysis seem to be helpful to the enrichment of ascorbic acid (AsA) and fatty acids in seabuckthorn fruit, among which GalLDH, GMPase, ACC and TER are the potential main pathogenic genes to control the contents of AsA and fatty acids in fruit, respectively. This paper provides new insights for the molecular basis of chemical innovation of Hippophae rhamnoides L., and provides valuable resources for exploring the evolution and molecular breeding of Elaeagnaceae.
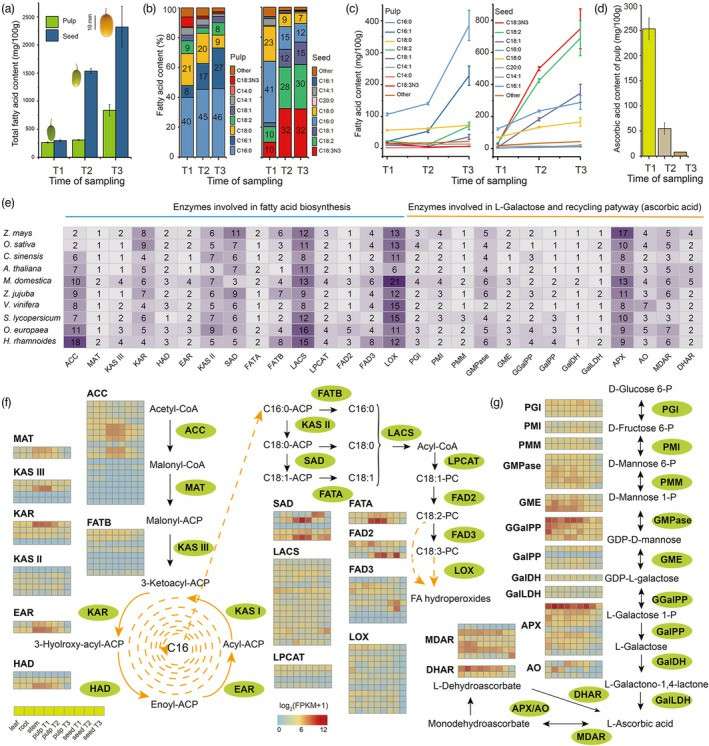 Content of fatty acid and ascorbic acid, and the expression of the related genes (Yu et al., 2022)
Content of fatty acid and ascorbic acid, and the expression of the related genes (Yu et al., 2022)
Pan-genome
In the study, 11 representative cucumber germplasm resources were selected, including 5 Indian wild groups, 2 East Asian groups, 3 European and American groups and 1 Xishuangbanna group. The genome assembly (Pacbio+10xGenomics+HiC) was completed at chromosome level, and the reference genome was added to construct cucumber pan-genome. By comparing these 12 genome sequences, it was found that there were 7 large DNA segment inversion variations on chromosomes 4, 5 and 7. These large inversion variations only existed in some wild materials, and the variations in different wild materials were different, which revealed the gradual evolution of inversion variation in the domestication of wild materials. The existence of inversion variation will lead to recombination inhibition and further limit the mining and utilization of wild resource genes.
The evolution history of inversion variation of these extra-large fragments provides important information for the selection and application of wild materials. In order to further reveal the genetic variation affecting cucumber domestication and important agronomic traits by using the graphic structure pan-genome, the characteristics of cucumber fruit thorn, flowering time and root development were deeply analyzed. A new 51-bp structural variation was found on the exon of CsTu, a key gene of fruit tumor, which can destroy the function of CsTu gene and affect fruit tumor. The genetic variation of multiple genes of fruit thorn tumor in 115 cucumber core collections was systematically analyzed, revealing the domestication and breeding selection process of structural variation in genes related to fruit thorn tumor traits.
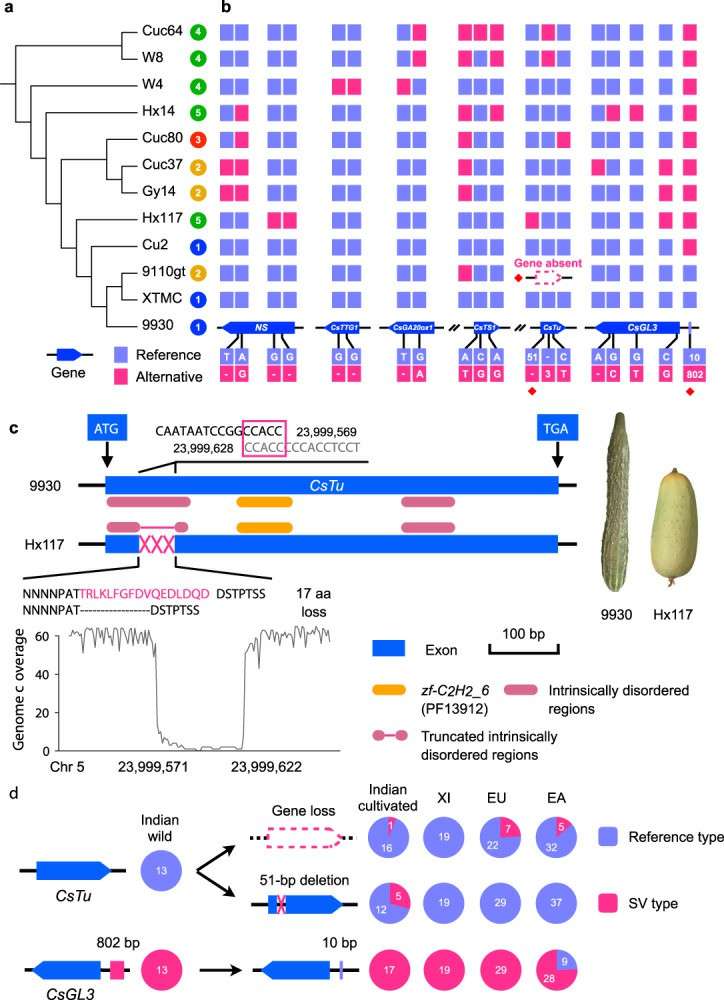 Allelic variants in genes involved in cucumber fruit spine and wart development (Li et al., 2022)
Allelic variants in genes involved in cucumber fruit spine and wart development (Li et al., 2022)
T2T Genome
Five of the 11 chromosomes of banana were assembled from telomere to telomere by using long-length Nanopore sequencing. A long-scattered repetitive sequence (LINE) named Nanica can be clearly identified on each chromosome of the newly assembled genome, which has been confirmed to exist in the centromere region by cytogenetic analysis. In addition, the 5S rDNA tandem repeats co-located with the centromere Nanica cluster were also clearly identified. This study also found many tandem repeats containing important gene families such as terpenoid synthase or disease resistance genes, which provided important resources for finding genes resistant to devastating diseases of banana crops such as black leaf spot. This high-resolution centromere region provided by the nearly complete genome of banana opens up a new way to study how satellite repeats are generated and evolved in the centromere region.
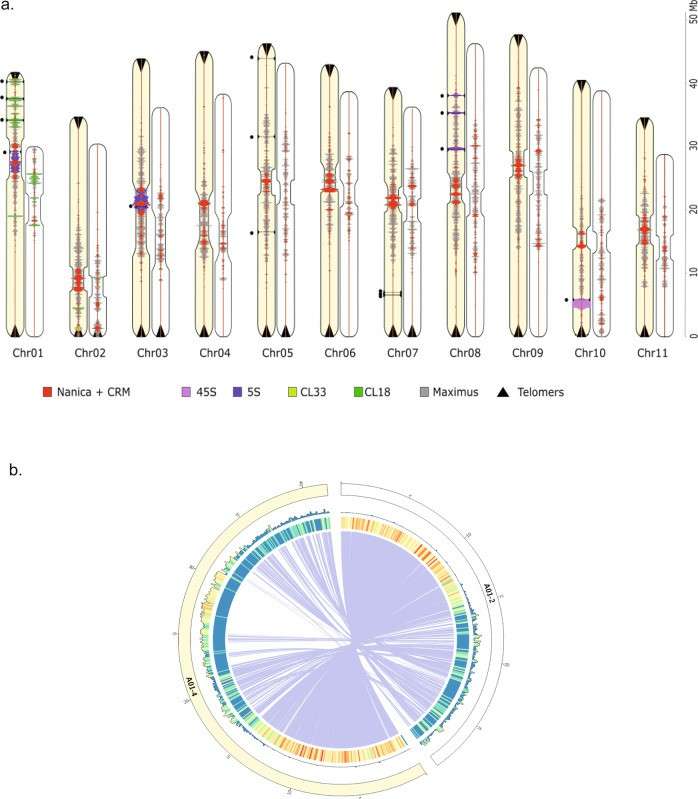 Comparison of the V2 and V4 assemblies (Belser et al., 2021)
Comparison of the V2 and V4 assemblies (Belser et al., 2021)
Haploid/Sub-genome and Transcriptome
Litchi chinensis belongs to Sapindaceae and is a tropical fruit with unique flavor. Feizixiao is one of the widely cultivated litchi varieties, and its genome is highly heterozygous (~2.27%). In this study, two sets of high-quality "Feizixiao" haplotype genomes (Size 470 Mb) were constructed by techniques such as Ilumina, PacBio, Hi-C and 10X. It was found that 13517 alleles were differentially expressed in different tissues. The resequencing analysis of 72 litchi materials showed that litchi had two independent domestication events. The very early-maturing litchi varieties domesticated in a wild population from Yunnan mainly correspond to one of the haplotypes of "Feizixiao". However, the late-maturing litchi variety domesticated independently in a wild population from Hainan corresponds to another haplotype. Early-maturing litchi varieties may be formed by crossing extremely early-maturing litchi varieties with late-maturing litchi varieties and cultivated in Guangdong.
A total of 13,517 differentially expressed alleles (DEA) were identified from 39 samples of different tissues and periods of princess smiles. Only 178 genes always prefer to express one allele, and a large number of genes are differentially expressed in different tissues. The distribution of DEA in the genome is not uniform, and it is concentrated in some areas, which may be related to specific biological processes. The research team also found that DEA and EEA have the same Ks value, but DEA suffers from stronger selection pressure. DEA has higher SNP density than EEA in Promoter, 5'UTR, intron and 3'UTR, but the SNP density of DEA in exon is significantly lower than that of EEA, which indicates that the two alleles of DEA have their own functions and are less tolerant of mutation.
However, the two alleles of EEA are similar or identical in function, and have certain substitution with each other, so they are more tolerant of mutation. It was found that the deletion variation of 3.7kb sequence containing a pair of CO-like genes may regulate the difference of fruit maturity among different litchi varieties. This study provides a new perspective for the domestication history of litchi, and also provides valuable resources for accelerating the breeding and improvement of litchi and related crops.
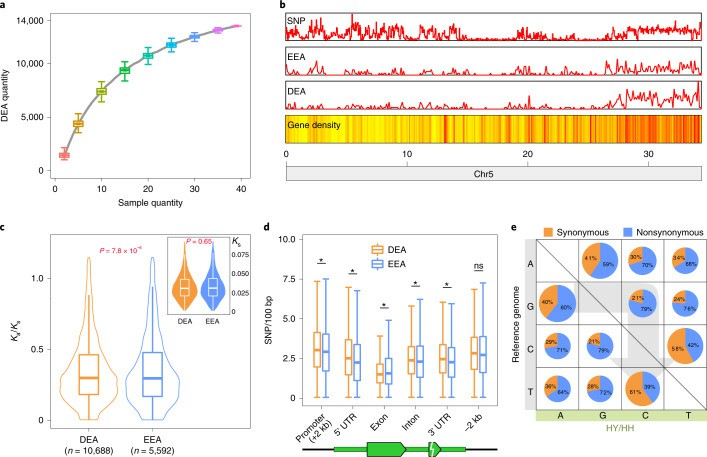 DEAs in lychee (Hu et al., 2022)
DEAs in lychee (Hu et al., 2022)
References
- Zhang Y, Yu Z., et al. "Comparative Genomics Reveals Evolutionary Drivers of Sessile Life and Left-right Shell Asymmetry in Bivalves." Genomics Proteomics Bioinformatics. 2022 20(6):1078-1091 https://doi.org/10.1016/j.gpb.2021.10.005
- Groppi A, Decroocq V., et al. "Population genomics of apricots unravels domestication history and adaptive events." Nat Commun. 2021 12(1):3956 https://doi.org/10.1038/s41467-021-24283-6
- Lin P, Yin H., et al. "The genome of oil-Camellia and population genomics analysis provide insights into seed oil domestication." Genome Biol. 2022 23(1):14 https://doi.org/10.1186/s13059-021-02599-2
- Yu L, Zhang J., et al. "Genome sequence and population genomics provide insights into chromosomal evolution and phytochemical innovation of Hippophae rhamnoides." Plant Biotechnol J. 2022 20(7):1257-1273 https://doi.org/10.1111/pbi.13802
- Li H, Zhang Z., et al. "Graph-based pan-genome reveals structural and sequence variations related to agronomic traits and domestication in cucumber." Nat Commun. 2022 13(1):682 https://doi.org/10.1038/s41467-022-28362-0
- Belser C, Aury JM., et al. "Telomere-to-telomere gapless chromosomes of banana using nanopore sequencing." Commun Biol. 2021 4(1):1047 https://doi.org/10.1038/s42003-021-02559-3
- Hu G, Li J. Two divergent haplotypes from a highly heterozygous lychee genome suggest independent domestication events for early and late-maturing cultivars. Nat Genet. 2022 54(1):73-83 https://doi.org/10.1038/s41588-021-00971-3


 Send a Message
Send a MessageFor any general inquiries, please fill out the form below.


