Mapping m6A on Viral RNA in Infected Cells: When to Use SELECT, MeRIP-qPCR, or Transcriptome-Wide Sequencing
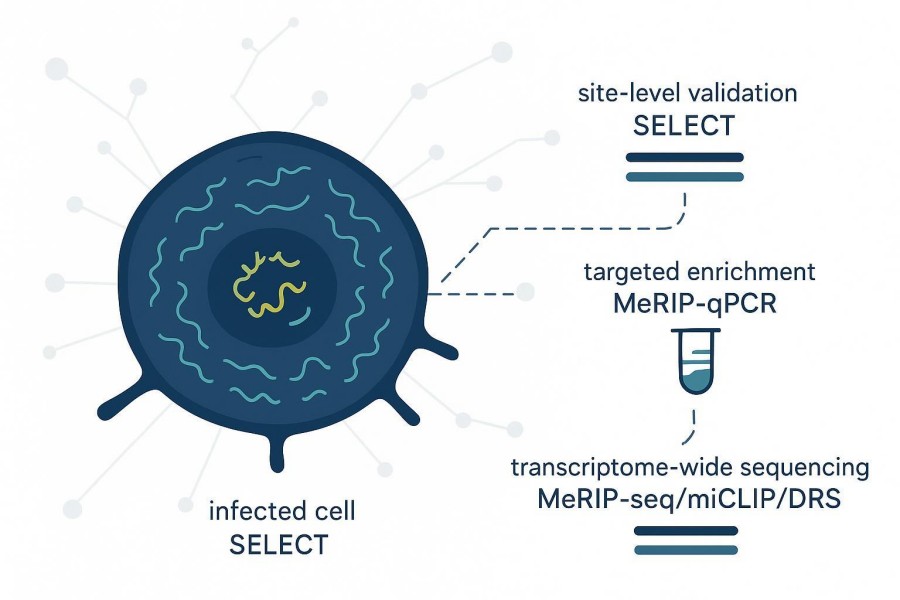
Viral RNA m6A mapping in infected cells is less about which technology sounds most comprehensive and more about what your result must prove inside a mixed host-viral RNA background. If your goal is single-site confirmation, SELECT is purpose-built. If you need regional enrichment comparison at one gene or a small target set, MeRIP-qPCR fits. If you need broader context across viral and host transcripts, transcriptome-wide sequencing (for example, MeRIP-seq peaks, miCLIP single-nucleotide maps, or nanopore direct RNA sequencing) is the right scale. In other words, choose the method that matches the claim you actually need to support-especially when infected-cell material makes viral signal compete with abundant host RNA. This article focuses on method fit and interpretation logic rather than declaring a universal winner, and it uses recent evidence to ground each recommendation. If you need a broader primer on the field, start with CD Genomics' overview of m6A detection methods, and for a direct mapping-method comparison see MeRIP-seq vs GLORI vs CAM-seq for m6A mapping. To use this guide efficiently, start with the claim you need to support (single site, target-region enrichment, or transcriptome-wide context), then sanity-check your sample reality (especially how host-dominant the RNA background is), and finally pick the smallest method scope that can answer the question. Plan the "next-step trigger" in advance: what result would justify expanding from a targeted assay to broader sequencing, and what orthogonal validation you will run if signals disagree.
Key takeaways
- Start from the claim, not the tool. Pick the smallest method scope that still answers your question: site-level (SELECT), target-level enrichment (MeRIP-qPCR), or transcriptome-wide context (MeRIP-seq/miCLIP/nanopore DRS).
- In infected cells, the hard part is signal separation. Host RNA often dominates, so input strategy (total vs enriched viral RNA vs infected-cell lysate) and background controls shape confidence as much as the assay itself.
- Don't confuse abundance with modification. Enrichment signals (MeRIP-qPCR/MeRIP-seq) require matched input normalization and orthogonal checks; single-nucleotide claims demand site-level validation.
- Stage your work. Many strong studies start narrow (SELECT or MeRIP-qPCR) and expand only when early results justify transcriptome-wide profiling.
- Plan for interpretation. Decide in advance what would count as success (validated site, target-level enrichment change, or landscape context) and which follow-ups you'll run if you see conflicting signals.
In infected cells the main design problem is signal separation for viral RNA m6A mapping
Infected-cell samples rarely present viral RNA in isolation. Instead, they are complex mixtures in which host RNA typically outweighs viral transcripts by orders of magnitude. That imbalance means method choice is as much about handling background as it is about detecting m6A-the right fit depends on how cleanly you can separate viral-specific signal from everything else and the granularity of evidence your study needs. Reviews of viral m6A biology emphasize that infected-cell context complicates interpretation and often requires multi-strategy workflows and careful controls to avoid overclaiming site specificity or stoichiometry, as summarized in 2024 perspectives on infection studies.
Viral RNA and host RNA compete in the same sample context
In host-dominant samples, viral reads can be sparse, which weakens statistical power for broad profiling and increases the chance that antibody-based enrichments or model-based calls reflect noise or mapping artifacts rather than bona fide modification. Practical protocol guidance for infected-cell MeRIP experiments underscores matched input controls, antibody validation, and fragmentation to ~100-200 nt to manage specificity and resolution, principles that carry directly to viral systems, as shown in a SARS-CoV-2 MeRIP-Seq protocol from 2021 (Li and Rana, STAR Protocols 2021). Recent viral-context reviews also advise orthogonal validation when background is high and viral load is low (Horner 2024 review of infection studies).
The same m6A question can imply different goals
"Is this viral site m6A-modified?" is not the same question as "Does a region on this viral transcript show higher enrichment under condition A than B?" or "What does the overall viral-host modification landscape look like during infection?" Each requires a different method and supports a different kind of claim. Site-level confirmation (SELECT) can directly address a narrow hypothesis. Target-focused enrichment (MeRIP-qPCR) helps compare specific viral regions across conditions. Transcriptome-wide mapping (MeRIP-seq peaks, miCLIP single-nucleotide events, or nanopore DRS site calls) is stronger when you need discovery, distribution patterns, or integration with host context.
Start method choice from resolution needs
Think in three tiers of resolution: single nucleotide, regional enrichment, and transcriptome landscape. SELECT is designed for the first tier. MeRIP-qPCR yields regional comparisons at a small set of targets. MeRIP-seq, miCLIP, and nanopore DRS provide the broader landscape with very different trade-offs: MeRIP-seq identifies ~100-200 nt peaks (Ge et al., Nat Commun 2022); miCLIP can localize to single bases with crosslink signatures (Linder et al., Nat Methods 2015); DRS works on native RNA with model-dependent calls that are coverage- and tool-sensitive (Parker et al., eLife 2020; Maestri et al., Brief Bioinform 2024).
 Suggested caption: In infected-cell samples, method choice depends on how much viral-specific resolution is needed within a mixed host-viral RNA background.
Suggested caption: In infected-cell samples, method choice depends on how much viral-specific resolution is needed within a mixed host-viral RNA background.
Start by asking what the result must prove
Before discussing kits or pipelines, define the proof you need. Are you trying to confirm a single predefined site? Compare enrichment at one viral gene across conditions? Or characterize the broader modification landscape across viral and host transcripts? Your first method should be the smallest one that answers that exact question.
For one predefined site or a very small number of targets
SELECT is strongest when your hypothesis is narrow and site-focused. It's a sequencing-independent assay that detects modification at a specific nucleotide via ligation/extension behavior captured by qPCR readouts. Recent reviews outline how SELECT can validate specific sites identified by broader screens, provided that primers avoid problematic secondary structures and appropriate controls are included (e.g., input normalization and, when feasible, writer perturbation) (Körtel et al., NAR 2021; Yang et al., Front Cell Dev Biol 2024). This single-site emphasis makes SELECT well-suited to infected-cell projects that value a crisp, interpretable result at a candidate locus.
For comparing enrichment at one gene or a small target set
MeRIP-qPCR is useful when you already have candidate viral regions or transcripts and need targeted enrichment comparisons under matched input and, ideally, IgG control. In infected-cell contexts, it offers a practical middle path: strong enough to quantify pull-down differences at predefined regions without the cost and complexity of full transcriptome mapping, but not intended for single-nucleotide localization. Viral studies have successfully used MeRIP followed by qPCR to assess specific genomic elements under perturbations, with the caveat that enrichment must be interpreted alongside RNA abundance-for example, m6A on HBV pgRNA epsilon regions reported with IP/qPCR comparisons (Hou et al., PNAS 2018).
For broader context across viral and host transcripts
If your questions are discovery-oriented-where are the enriched regions; which transcripts change most; how does the viral landscape compare to host-then transcriptome-wide sequencing is justified. MeRIP-seq maps peak-level enrichment across the transcriptome (~100-200 nt regions), miCLIP can localize to single nucleotides using crosslink signatures, and nanopore direct RNA sequencing (DRS) examines native RNA with model-based site calls that depend strongly on coverage and tool selection. Each approach offers wider context and hypothesis generation, but each also raises distinct control and validation needs, as highlighted in benchmarking and reviews (Ge 2022; Parker 2020; Tan et al., mSystems 2024; Maestri 2024).
| Study goal | Best-fit method | Why it fits | What it does not resolve well | Typical follow-up | Representative refs |
|---|---|---|---|---|---|
| Confirm a predefined single site | SELECT | Site-level, single-nucleotide confirmation with a qPCR readout and targeted primers | Regional patterns, discovery across other transcripts | Expand to MeRIP-qPCR for regional comparison; add writer perturbation or orthogonal checks | Körtel 2021; Yang 2024 |
| Compare enrichment on 1-3 viral targets | MeRIP-qPCR | Quantifies IP enrichment vs input/IgG across defined regions | Single-base precision; confounding by expression changes without careful normalization | Validate key candidate sites by SELECT; consider expanding to MeRIP-seq for broader context | Li & Rana 2021; Hou 2018 |
| Discover or contextualize viral and host patterns | MeRIP-seq/miCLIP/DRS | Transcriptome-wide mapping for landscape and candidate discovery | Immediate site-level proof (MeRIP-seq); tool- and coverage-dependent calls (DRS); higher complexity | Validate priority sites by SELECT or miCLIP; integrate matched input RNA-seq for MeRIP comparisons | Ge 2022; Parker 2020; Maestri 2024 |
SELECT, MeRIP-qPCR, and transcriptome-wide sequencing answer different questions
Treat these methods as complementary lenses rather than competitors. They differ most in resolution, susceptibility to background, and the type of claim they can credibly support.
When SELECT is the strongest choice
Choose SELECT when you already have a narrowly defined site-level hypothesis and need direct, nucleotide-specific evidence. SELECT's qPCR-based readout focuses interpretation on a single locus, which is ideal when infected-cell background might otherwise blur region-level signals. Good practice includes careful primer design around structure, technical replicates for the ΔCt-like signal, and including controls that strengthen causal interpretation (e.g., writer knockdown where feasible). SELECT is not intended to scan entire transcripts or discover new candidate regions; it is a validation tool for focused questions (see Körtel 2021 and Yang 2024 for design and controls).
When MeRIP-qPCR is the stronger middle path
Pick MeRIP-qPCR when you want to compare enrichment at one gene or a few viral targets across conditions without committing to full transcriptome profiling. It measures antibody IP relative to matched input (and often IgG), providing interpretable fold-enrichment metrics at predefined regions. In host-dominant samples, MeRIP-qPCR can be more robust than trying to power a transcriptome-wide experiment that may be under-sampled on viral reads. The trade-off is resolution: you'll see region-level changes, not single-nucleotide events, and you must interpret any enrichment changes in the context of underlying expression (see Li & Rana 2021 protocol notes and viral exemplars like Hou 2018).
When transcriptome-wide sequencing is justified
Use transcriptome-wide approaches when you need discovery or landscape-scale context that smaller assays cannot provide. MeRIP-seq offers peak-level maps across viral and host RNAs, which is useful for pattern recognition and candidate nomination, but it cannot by itself prove site-level changes and depends on matched input RNA-seq for interpretation (Ge 2022). miCLIP increases resolution to single nucleotides but demands higher library and computational rigor (Linder 2015). Nanopore DRS works on native RNA, potentially revealing isoform context and full-length patterns; however, modification calls are model-driven, coverage-sensitive, and benefit from stringent thresholds and orthogonal validation to manage false positives in mixed backgrounds (Parker 2020; Tan 2024; Maestri 2024).
Why these methods are often staged rather than exclusive
Many strong programs start narrow and expand only when the first answer justifies it. A common pattern is to use MeRIP-qPCR to compare a handful of viral regions or conditions, validate a few compelling sites with SELECT, and then move into MeRIP-seq or DRS to contextualize those hits across viral and host transcripts. This sequence respects both budget and interpretability by ensuring that broader profiling is anchored to validated biology rather than driven by habit (see background cautions in Horner 2024 and power/replicate discussions around MeRIP analyses).
 Suggested caption: SELECT, MeRIP-qPCR, and broader sequencing differ most in resolution, scope, and the type of claim they can support.
Suggested caption: SELECT, MeRIP-qPCR, and broader sequencing differ most in resolution, scope, and the type of claim they can support.
The sample strategy matters as much as the method
A minimal viability checklist before you pick a method
- Define the claim tier up front: site-level confirmation, region-level enrichment comparison, or transcriptome-wide landscape/discovery.
- Lock the background context: infection model and timepoint, input type (total vs enriched viral RNA vs lysate), and an estimated viral:host RNA ratio (even a rough estimate helps).
- Pre-specify the controls you can support: matched input is essential for enrichment-based methods; include an IgG control when feasible; consider writer/eraser perturbation or orthogonal validation for any site-level claim.
- Set a reproducibility minimum: prioritize biological replicates; treat technical replicates as readout-stability checks (especially for qPCR-based assays).
- Record what reviewers will ask for later: RNA integrity/QC metrics, fragmentation or library choices, mapping/reference details, and any enrichment history so interpretation remains traceable.
Two projects using the same assay can reach different levels of confidence if their input strategies differ. Total RNA from infected cells preserves host-dominant background, which reduces the viral-specific signal available to any method. Enriching viral RNA (for example, capture of viral sequences) can sharpen signals for both targeted and broad assays, and documenting infection timing, input QC, and enrichment history helps reviewers and collaborators interpret your results.
For a broader discussion of why viral RNA modification readouts can be tricky to interpret in infection models (and how measurement choices affect the story you can tell), see viral RNA modification and mRNA vaccine research measurement strategies.
Input types shape analytical context
- Total RNA from infected cells: maximally realistic but host-heavy; appropriate when you intend to model the full cellular context or when enrichment is not feasible.
- Enriched viral RNA: increases viral:host ratio, potentially improving detection power for MeRIP-qPCR and transcriptome-wide profiling; requires clear documentation to avoid misinterpretation of relative abundance changes.
- Direct infected-cell input (e.g., IP from lysate): convenient, yet background and nonspecific binding can rise; matched input and IgG controls become more critical.
How host background alters viral-specific claims
When viral RNA makes up a small fraction of total reads, transcriptome-wide peak calling becomes sensitive to replicate number and depth, and DRS site calls become tool- and threshold-dependent. In these cases, small, targeted assays can offer cleaner answers. Conversely, when infections are robust and viral RNA is abundant, landscape-scale methods become more powerful and reproducible. The choice is not ideological-it's about signal-to-noise under your specific sample realities. Replicate-aware analyses of MeRIP data demonstrate that statistical power grows with depth and replicates, reducing false discoveries under noisy backgrounds (see differential MeRIP modeling in RADAR 2019).
Why traceable input history improves selection
Method selection benefits from a traceable record: infection model and timepoint, input type (total, enriched, lysate), RNA integrity, approximate viral:host ratio, and how many replicates and controls you can support. That context allows you to scope the first method realistically and to specify what triggers an expansion step if early signals suggest broader questions.
What usually makes viral RNA m6A results hard to interpret
Misinterpretation typically arises from conflating evidence layers and underestimating the effect of host background on each method's readout. Keep three layers distinct in your mind and in your report: RNA abundance (expression), enrichment-based signal, and site-level validation.
Expression change is not enrichment change
If viral RNA abundance changes across conditions, enrichment signals can shift even if modification levels do not. Matched input normalization and, when feasible, perturbation controls that specifically affect the modification machinery help separate abundance effects from modification effects. Analyses of MeRIP-based workflows note that replicate number and depth strongly affect peak detection power; under-sampled designs can overstate changes or miss them altogether (McIntyre et al., Sci Reports 2020).
Host-dominant samples weaken viral-specific confidence
With host >> viral RNA, antibody pull-downs and model-based calls have more background to contend with. MeRIP-seq peak calling becomes noisier unless you increase depth and replicates, and DRS site detection becomes highly sensitive to coverage thresholds and the choice of computational tools. In these situations, start with focused questions and escalate only when early findings are robust, as advised in recent viral DRS benchmarking (Tan 2024) and tool-comparison studies (Maestri 2024).
Site-level and region-level claims are not equivalent
A site-level claim requires a site-level method. SELECT or miCLIP can provide nucleotide resolution; MeRIP-qPCR and MeRIP-seq cannot. Use precise language in your write-ups so readers do not interpret regional enrichment as proof of a specific modified adenine (principles summarized across Ge 2022 and Linder 2015).
Broad sequencing does not answer a focused mechanism by itself
Transcriptome-wide data add context and can reveal unexpected candidates, but they do not automatically deliver the most relevant answer for a narrow mechanism. Plan orthogonal validation for any high-priority sites and treat discovery maps as the starting point for targeted testing, not the finish line (see general DRS guidance in Parker 2020).
 Suggested caption: In infected-cell studies, abundance change, enrichment change, and site-level confirmation are different layers of evidence.
Suggested caption: In infected-cell studies, abundance change, enrichment change, and site-level confirmation are different layers of evidence.
A practical path to method selection for infected-cell viral RNA
Here's a pragmatic way to decide, designed for host-dominant, real-world samples: start with the narrowest method that still answers your question; define what success looks like; and state what would trigger a staged expansion.
Start with the narrowest claim you actually need
If your biological question can be answered by confirming a single site, do that first. It protects interpretability and budget while giving you a clear go/no-go result. Think of it this way: when the signal is faint and the background loud, a spotlight works better than a floodlight.
Use targeted methods when a target is defined
When you have one or a few viral regions of interest, targeted enrichment comparison via MeRIP-qPCR can deliver fast, interpretable answers-provided you normalize to matched input and include appropriate controls. If one region stands out, validate the most compelling site(s) with SELECT before scaling up.
Move to broader sequencing only when required
Expand to transcriptome-wide mapping when discovery or broader context truly matters: for example, when early targeted results suggest widespread changes, when host-viral comparisons are central to your hypothesis, or when you need to evaluate isoform-level patterns. For MeRIP-seq, plan for matched input RNA-seq and adequate replicates; for DRS, ensure coverage and select tools/cutoffs conservatively, anticipating orthogonal validation.
Decide early what follow-up counts as success
Success might be "we confirmed site X" (SELECT), "we observed consistent enrichment shifts at the target region across replicates" (MeRIP-qPCR), or "we mapped a viral-host landscape and nominated candidates for validation" (transcriptome-wide). Write these outcomes down in advance and note the trigger conditions for moving from one stage to the next.
| Project situation | First method to consider | Why | What may trigger expansion | Most likely next step | Representative refs |
|---|---|---|---|---|---|
| Single predefined site, low viral burden | SELECT | Site-level proof with clear interpretation under background | Additional hypotheses emerge; need region/landscape context | MeRIP-qPCR on nearby regions; later MeRIP-seq/DRS for context | Körtel 2021 |
| A few viral targets, need condition comparison | MeRIP-qPCR | Targeted enrichment vs input/IgG; faster iteration than full profiling | Unexpected off-target signals; inconsistent enrichment across targets | SELECT on top sites; expand to MeRIP-seq for broader survey | Li & Rana 2021; Hou 2018 |
| Discovery or host-viral context is central | MeRIP-seq/miCLIP/DRS | Landscape mapping and candidate nomination | Priority candidates identified; need site-level proof | SELECT/miCLIP validation at key loci; refine DRS thresholds/tools | Ge 2022; Maestri 2024 |
What good deliverables look like for a viral RNA m6A project
Good deliverables make interpretation straightforward and reanalysis possible. They should document inputs, controls, QC metrics, analysis outputs, and clear caveats about what each method can and cannot prove in infected-cell material.
For SELECT-like projects
Expect a crisp definition of the target site(s), primer design notes (including secondary-structure considerations), technical replicate handling, and a clear table or plot of the site-level readout across conditions. Interpretation notes should specify controls (e.g., input normalization; any writer perturbation) and explain exactly what the ΔCt-like signal represents in your design. If you plan staged work, the deliverable should also suggest which neighboring regions or orthogonal methods (e.g., MeRIP-qPCR) would be the next logical step, reflecting best-practice validation sequences highlighted in 2021-2024 reviews.
For MeRIP-qPCR projects
A strong package includes raw Ct values for input, IP, and optional IgG; %Input and fold-enrichment calculations; matched input normalization rationale; and bar charts or comparable visualizations with error bars across replicates. The report should annotate target regions on viral transcripts and state limitations clearly (regional resolution; expression-enrichment disentanglement). As an example of expected components, see how service descriptions enumerate input/IgG controls, %Input and Fold Enrichment calculations, and QC summaries in a typical report.
For transcriptome-wide projects
For MeRIP-seq, expect a QC section (data quality control, fragment-length distribution, library metrics), peak statistics (number, width, genomic/transcriptomic distribution), motif enrichment, and coverage/annotation tracks that are ready to inspect in genome browsers. Matched input RNA-seq summaries should be included to contextualize enrichment changes. For DRS, expect coverage summaries per viral and host transcript, the toolchain and thresholds used for site calling, and a prioritized candidate list with suggested orthogonal validations. These deliverables should be reproducible and analysis-ready (for example, FastQ/BAM files, peak tables, motif results, QC reports) so you can integrate them into follow-up experiments.
If you want a concrete sense of what "good deliverables" look like in practice, the following research-use-only (RUO) pages can be used as neutral examples of what to ask for in a final report (controls documented, QC summaries, and analysis-ready files):
- SELECT m6A site validation deliverables (RUO)
- MeRIP qPCR enrichment reporting and controls (RUO)
- MeRIP seq QC modules and analysis outputs (RUO)
Disclosure: Links above are provided for deliverable/QC expectations and scoping reference; they do not change the method-selection logic in this article.
FAQs about viral RNA m6A mapping in infected cells
Which method is best for validating m6A on viral RNA in infected cells?
It depends on what you must prove. Use SELECT when you already have a narrow, site-level hypothesis and want nucleotide-specific evidence; use MeRIP-qPCR when you need targeted enrichment comparisons at one gene or a small set; and move to transcriptome-wide approaches (MeRIP-seq peaks, miCLIP single-nucleotide maps, or nanopore DRS) when discovery or broader host-viral context matters most. Recent reviews describe SELECT as a reliable single-site validator, MeRIP workflows as regional enrichment tools that require input normalization, and DRS as powerful but model- and coverage-dependent-each with distinct validation needs (Yang 2024; Ge 2022; Parker 2020).
Can MeRIP-qPCR distinguish viral RNA m6A change from expression change by itself?
Not completely. MeRIP-qPCR quantifies immunoprecipitation relative to matched input (and often IgG), so enrichment can shift if expression shifts. To separate abundance from modification effects, interpret enrichment alongside RNA abundance and, when feasible, use orthogonal validation such as SELECT at key sites or writer perturbation experiments suggested in contemporary protocols and reviews (Li & Rana 2021; McIntyre 2020).
Is transcriptome-wide sequencing always better because it gives more data?
No. More data do not necessarily produce the most relevant answer for a focused mechanism. MeRIP-seq identifies ~100-200 nt enrichment regions that are excellent for landscape and candidate nomination but not for immediate site-level proof. Nanopore DRS yields model-based site calls that depend on coverage and tool thresholds and generally benefit from orthogonal validation. Use broad profiling when context or discovery is central to your hypothesis, and plan targeted follow-ups in advance (Ge 2022; Maestri 2024).
When is SELECT a better choice than MeRIP-qPCR?
When your project has a predefined single-site hypothesis and you need nucleotide-level evidence, SELECT is the more direct route. MeRIP-qPCR is stronger when you need to compare enrichment across a defined region or a handful of targets but do not require single-nucleotide localization or transcriptome-wide context yet (Körtel 2021; Yang 2024).
What is the biggest reason viral RNA m6A studies in infected cells become hard to interpret?
Usually a mismatch between method resolution, host-background complexity, and the strength of the claim. For example, reading regional enrichment as if it were site-specific evidence, or relying on under-powered transcriptome-wide data without matched input normalization and orthogonal validation, can lead to overconfident conclusions. Define the claim first, pick the appropriate method, and plan follow-ups that address the known limitations of each approach (see cautions in Horner 2024 and replicate/power considerations in McIntyre 2020).
When the project is ready to scope
Projects tend to move faster and produce clearer conclusions when a few readiness signals are present. Rather than listing a long checklist, look for these concise indicators and blockers.
You're likely ready when your target question is crisp (site-level vs target-level vs landscape), the sample context is defined (infection model and timepoint, input type, expected viral:host ratio, and planned controls), the level of evidence you need is explicit, and the team agrees on the first output and follow-up plan. If the question remains broad, input strategy is unclear for a host-dominant sample, the team expects one method to answer every level of question, or background effects have not been considered in replicate/power planning, refine the design first. Before requesting technical input, prepare your viral system, infection model and timepoint, input type (total vs enriched vs lysate), target or discovery goal, approximate sample number and replicates, and whether staged follow-up is acceptable. This summary lets collaborators or service teams scope a realistic first method and define reliable triggers for expansion.
If you anticipate integrating viral m6A mapping with other readouts (expression, chromatin, or additional RNA modifications), a practical next read is experimental design for multi-omic epitranscriptomics and RNA-modification crosstalk.
Selected references and further reading
- Protocol for infected-cell MeRIP with matched input and antibody validation: Li & Rana, STAR Protocols 2021.
- Site-level validation logic for SELECT: Körtel et al., Nucleic Acids Research 2021 and Yang et al., Frontiers in Cell and Developmental Biology 2024.
- Peak width and landscape context for MeRIP-seq; single-nucleotide mapping by miCLIP: Ge et al., Nat Commun 2022 and Linder et al., Nat Methods 2015.
- DRS principles and benchmarking under coverage/tool constraints: Parker et al., eLife 2020; Tan et al., mSystems 2024; Maestri et al., Briefings in Bioinformatics 2024.
- Viral-context interpretation cautions in infected-cell studies: Horner 2024 review.




