Whole Blood, Plasma, or Extracted cfDNA for Methylation Studies? A Pre-Analytical Guide
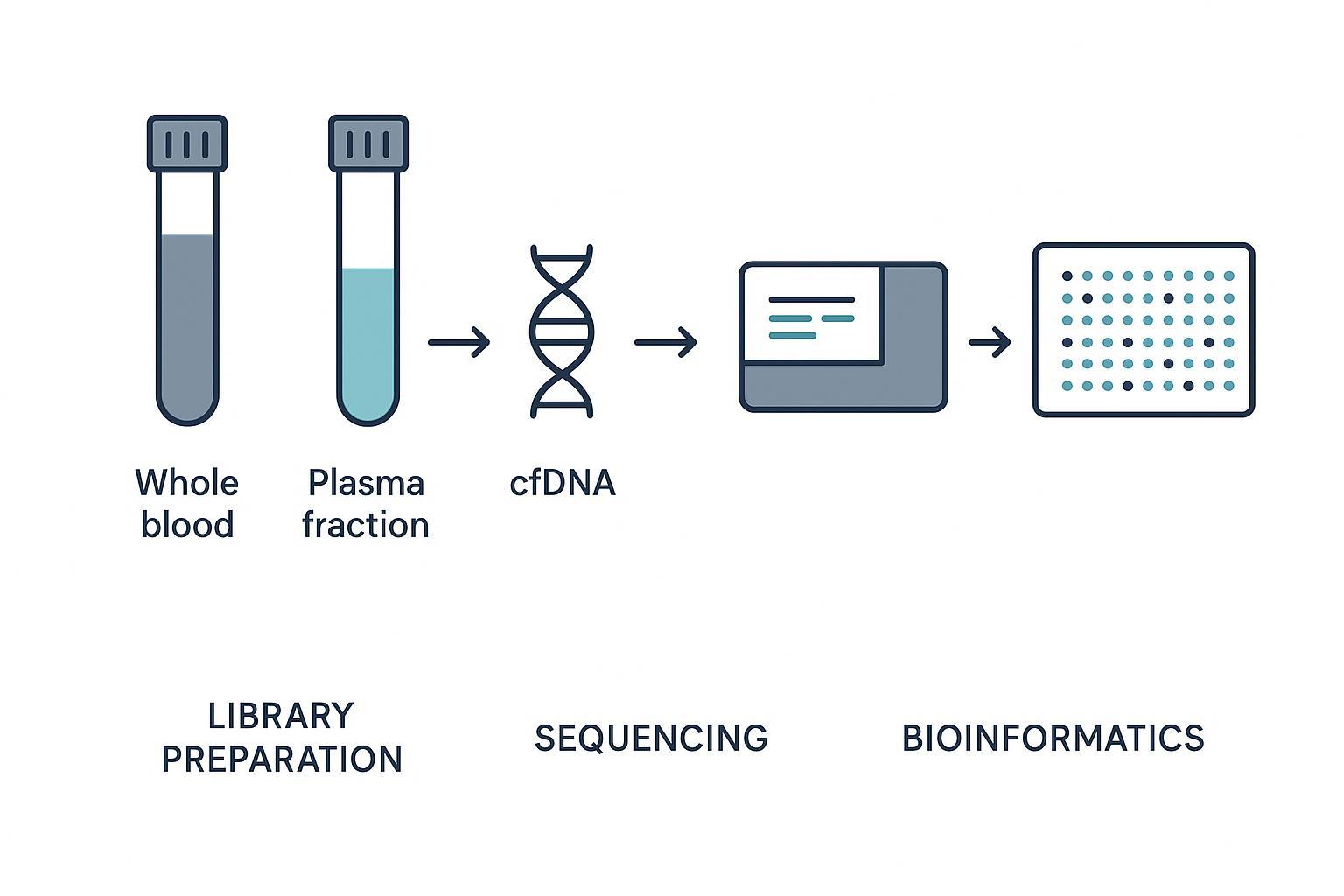
There is no single best sample type for every project. The right choice depends on how much control you have over handling, how consistently you can apply the workflow across sites, and how traceable your sample history will be from collection to analysis. In other words, cfDNA methylation sample requirements are not one-size-fits-all-and treating "whole blood vs plasma cfDNA methylation" as a universal either-or misses the operational reality. This guide is a practical cfDNA sample type guide that shows how to choose between whole blood, plasma, and extracted cfDNA by balancing pre-analytical risk, interpretability, and quote planning.
Key takeaways
- Whole blood, plasma, and extracted cfDNA are all viable submission formats-but they are not interchangeable. Base your choice on control, consistency, and traceability rather than theory alone.
- Plasma often provides a cfDNA-enriched input with less background genomic DNA when separation and storage are consistent and well documented.
- Whole blood centralizes processing under a single SOP when sites can't reliably separate plasma; it requires planning to control delays and transport.
- Extracted cfDNA can accelerate project launch only when extraction history and QC records are complete; without documentation, scope reliability and interpretability suffer.
- Quote quality improves when teams share clear submission format, cohort structure, handling history, and analysis goals up front; this reduces rescoping later.
Which Sample Type Is Best for a cfDNA Methylation Project?
Short answer: none of them, universally. Each format shifts the balance between convenience, upstream control, traceability, and downstream interpretation. If you prioritize cross-site/batch consistency, you may reach a different conclusion than a team that prioritizes local convenience or immediate startup.
Why this decision matters more than many teams expect
Submission format influences background DNA, handling variability, extraction history, and ultimately how confident you can be in methylation comparisons. Reviews and harmonization guidance stress that tube type, processing windows, and centrifugation parameters materially change the ratio of cfDNA to background leukocyte DNA in the analytical input, which in turn affects low-input methylation workflows and interpretability according to the NCI's evidence-based practices summarized in Greytak et al. (2020) in Harmonizing cell-free DNA collection and processing practices. See the detailed discussion in the NCI BEBP overview: NCI BEBP guidance on handling and tube effects (Greytak 2020).
Why this is not just a shipping question
Choosing a submission format is a design decision. It affects signal quality, pre-analytical risk, documentation burden, and even quote accuracy. Optimizing for "what's easiest to send today" can create downstream rework if handling history is incomplete, background DNA is high, or site-to-site drift isn't controlled.
 Different submission paths route into similar methylation workflows but impose different control, documentation, and risk profiles.
Different submission paths route into similar methylation workflows but impose different control, documentation, and risk profiles.
Whole Blood, Plasma, and Extracted cfDNA Each Solve a Different Problem
The practical question is not "which is best," but "which format aligns with your goals and operational constraints?" Think of the submission choice like choosing a lane on a highway: all lanes reach the destination, but traffic, visibility, and your ability to drive steadily vary.
When whole blood is the more practical option
Whole blood can be the right choice when you need a simpler collection workflow at distributed sites and want central processing to enforce a uniform SOP. If local teams cannot reliably separate plasma within the required timelines, shipping whole blood in stabilization tubes to a central lab can reduce site-to-site variability. This centralization shifts the risk from multiple local separations to a single, standardized process-especially useful in multi-site cohorts where comparability matters more than maximal theoretical purity. Evidence-based guidance notes that unprocessed blood is more sensitive to delays without stabilizers, while preservative tubes tolerate planned transit better than standard EDTA, as shown in Clin Cancer Research's evaluation of preservatives and temperature (Hyman 2017).
When plasma offers better upstream control
Plasma is commonly preferred when you can manage separation timelines, double-spins, and cold-chain storage consistently. By removing cells and platelets promptly and thoroughly, you reduce background leukocyte genomic DNA that can dilute cfDNA-specific methylation signals. Multiple studies demonstrate that two-step centrifugation and consistent handling lower background DNA and improve downstream analytics; for example, see paired processing recommendations and double-spin effects (Risberg 2018) and the broader harmonization perspective on processing windows (Greytak 2020).
When extracted cfDNA can simplify project launch
If you already have high-quality extracted cfDNA with complete metadata, submission can be fast and focused. The operative phrase is "with complete metadata": kit/protocol and version, operator and date, input volumes, elution buffers, fragment profile, concentration method, storage time/temperature, and freeze-thaw count. Without this information, it becomes difficult to assess input suitability or diagnose outliers downstream. Reviews synthesizing analytical-workflow risks emphasize that traceability enables reliable interpretation; see the recent overview of circulating nucleic acids workflow factors in Clinical Chemistry (van der Leest 2024) and the focused discussion of pre-analytical variables in Frontiers in Cell and Developmental Biology (Peng 2024).
Why "more processed" does not always mean "lower risk"
Pre-separated plasma or extracted cfDNA is not automatically more reliable. If separation or extraction was inconsistent, undocumented, or performed under variable conditions, you inherit invisible noise. In contrast, a centralized whole-blood route with a single, well-documented SOP may deliver more comparable inputs-especially when sites vary in capacity.
How Sample Type Changes Pre-Analytical Risk
Your submission format determines which pre-analytical risks you must manage most aggressively. Four stand out across the literature: background genomic DNA contamination, handling drift across sites/batches, storage and freeze-thaw effects, and traceability gaps.
Background genomic DNA contamination
Plasma prepared promptly and spun twice generally contains less background leukocyte DNA than serum or delayed whole blood, improving the effective signal for low-fraction methylation analyses. The risk hierarchy is consistent across reviews: standard EDTA blood requires short processing windows; preservative tubes stabilize signals during longer room-temperature periods; and thorough cell/platelet removal reduces carry-over. See the harmonized guidance in Greytak 2020 (NCI BEBP) and the comparative analysis of collection/processing effects in Risberg 2018.
Sample handling drift across sites or batches
Small differences in draw-to-spin time, centrifugation parameters, or storage create technical noise that can masquerade as biology. Harmonized SOPs and detailed metadata are the antidote. Document tube type and lot, draw time, processing times, g-forces and durations, temperatures, storage conditions, and chain-of-custody. The consistency theme recurs in both Greytak 2020's evidence-based recommendations and the 2024 overviews of circulating nucleic acid workflows in Clinical Chemistry (van der Leest 2024) and Frontiers in Cell and Developmental Biology (Peng 2024).
Storage, transport, and freeze-thaw effects
Cold-chain reliability matters. After separation, store plasma at -80°C, aliquot to minimize freeze-thaw cycles, and thaw on ice. Reviews note that repeated freeze-thaw cycles degrade cfDNA integrity and reduce yields; long-term storage at higher temperatures can alter analyte profiles. These practical recommendations are summarized in Clinical Chemistry's 2024 review of circulating nucleic acids workflows (van der Leest 2024) and expanded in Frontiers' analysis of pre-analytical variables (Peng 2024).
Why consistency often matters more than theoretical optimality
In real projects, a stable, traceable workflow usually beats a theoretically "purer" input created under inconsistent conditions. If you cannot keep separation timelines and double-spins uniform across sites, centralizing processing through a whole-blood route may yield more comparable datasets. Conversely, when you can enforce plasma SOPs locally, the cfDNA-enriched input can pay dividends in analysis.
 Before sequencing begins, sample type already influences background DNA, handling variability, storage sensitivity, and traceability.
Before sequencing begins, sample type already influences background DNA, handling variability, storage sensitivity, and traceability.
For a general background overview of cfDNA extraction materials, workflow, and sample handling, see this practical resource on cfDNA extraction and processing.
Which Submission Format Fits Different Research Scenarios?
Mapping submission format to real constraints helps avoid rework and keeps comparisons honest.
If your team needs the simplest collection workflow
When distributed collection sites have limited capacity for timed separations, whole blood can be easier to standardize. Specify tube types up front, align on transport and temperature control, and centralize separation under a single SOP. This approach concentrates risk management in one place and reduces the chance that subtle site differences creep into the data.
If your team wants tighter control over cfDNA-enriched input
When you can reliably meet separation timelines, document double-spin parameters, and enforce storage rules (aliquoting, -80°C, minimal thaw cycles), plasma is often the better fit. The payoff is lower background DNA and a clearer cfDNA signal, which supports more consistent methylation analysis provided documentation is thorough.
If your team already has extracted material ready
Submitting extracted cfDNA can jump-start a study when you've maintained meticulous records: protocol and version, input amounts, elution details, fragment profile, concentration method, and storage history. If any of those are missing, build a brief pilot or intake review to fill the gaps before scaling.
If your project compares multiple groups or collection sites
Standardization across all samples matters more than choosing the most technically attractive format for one subset. Pick the option you can execute uniformly, then commit to documenting it the same way for every sample and every batch.
How Sample Type Affects cfDNA Methylation Sample Requirements and Project Scope
Submission format shapes intake steps, QC gates, and the reliability of your quote. It also changes which details a provider must understand to model risk and plan capacity.
Why submission format changes service scope
In practice, each route concentrates different responsibilities. With whole blood, intake teams will verify tube types, transit conditions, and then apply a centralized separation SOP-trading many small upstream differences for one reproducible downstream process. With plasma, responsibilities shift upstream: local teams must record separation timings, double-spin parameters, and storage, because those factors govern background DNA. With extracted cfDNA, the focus narrows to extraction/QC documentation and shipment of small volumes under tight temperature control. The success of any route depends on how well these responsibilities are carried and documented.
What details a service provider usually needs before quoting
To keep scope stable and comparisons interpretable, it helps to send the same core fields for every sample and every batch. The table below can be pasted into an email or a sample manifest.
| Field to provide | Example (what "complete" looks like) | Why it matters for scope and interpretation | What goes wrong if it's missing |
|---|---|---|---|
| Submission format | Whole blood / plasma / extracted cfDNA | Determines upstream responsibilities, intake QC, and the main pre-analytical risks to model | Quote becomes conditional; results may be hard to compare across subsets |
| Species and matrix | Human plasma; mouse whole blood | Affects input expectations and reference resources | Rescoping after intake due to unsupported matrices or unclear background |
| Sample count and grouping | 40 samples: 20 vs 20; 2 sites; 2 batches | Enables planned contrasts and balanced batch strategy | Underpowered comparisons or batch/site confounding |
| Expected volume or yield | 4 mL plasma each; or ~5 ng cfDNA per sample | Drives library strategy options and contingency planning | Extra rounds of clarification; higher risk of late-stage exclusions |
| Tube type and handling window | EDTA; processed within same day; or stabilization tube with documented delay | Tube chemistry and delay strongly influence background gDNA | Unexplainable outliers; inconsistent background across sites |
| Centrifugation parameters (for plasma) | Two spins recorded with g-force, time, temperature | Defines how much cellular carry-over remains | Elevated background DNA; reduced interpretability of methylation differences |
| Storage and freeze-thaw history | Plasma at -80°C, aliquoted; 0-1 freeze-thaw | Storage drift can mimic biology and affects yield | Batch artifacts; lower yield; inconsistent fragment profiles |
| Extraction status + method (if applicable) | Extracted cfDNA: kit/protocol + version; date; input volume; elution details | Makes extraction history auditable and supports troubleshooting | "Black box" material: hard to diagnose failures or normalize across lots |
| QC snapshots (if available) | Concentration method; fragment profile trace; notes on HMW DNA | Accelerates intake triage and flags contamination early | Delayed start; late discovery of contamination or low complexity |
| Analysis goal (high-level) | Discovery screen vs targeted follow-up (research) | Prevents mismatch between study intent and deliverables | Misaligned scope and avoidable revisions |
This structure aligns with harmonization recommendations summarized in NCI BEBP's handling guidance (Greytak 2020) and expanded by Clinical Chemistry's 2024 workflow review (van der Leest 2024).
For a concrete example of how providers present these intake fields, review the sample requirements table on CD Genomics' cfDNA methylation/hydroxymethylation sequencing service (RUO) and mirror the same level of detail in your quote request.
If you're still early in planning, separate what you must provide to start scoping from what you should provide to avoid rescoping later:
| Planning stage | Minimum information to start scoping | Recommended information for a stable quote |
|---|---|---|
| Initial scope check | Submission format; species; sample count; high-level comparison; analysis goal | Adds tube type, processing window, storage temperature, and whether extraction is already done |
| Quote-ready brief | Adds expected volume/yield and basic handling notes | Adds full centrifugation parameters, freeze-thaw history, extraction protocol/version (if extracted), and QC snapshots (concentration + fragment profile) |
| Multi-site / multi-batch execution | Adds site list and a draft SOP alignment plan | Adds harmonized metadata schema, chain-of-custody fields, and a pre-defined deviation log for exceptions |
 A reliable quote starts with clear information about sample format, handling history, and study scope.
A reliable quote starts with clear information about sample format, handling history, and study scope.
Why incomplete sample information slows project planning
Gaps in handling history force teams to add contingencies: extra intake QC, holds for clarification, or even rescoping. Delays aside, incomplete documentation reduces interpretability, especially when technical artifacts can't be separated from biology due to unknowns in the workflow.
What makes a quote more reliable
Quotes improve when the submission format and cohort structure are locked early, intake details are thorough, and the analysis goal is explicit. In practice, this means you'll get a narrower, more stable estimate and fewer surprises if everything from tube type to freeze-thaw count is known ahead of time.
What to Send Along With the Sample
Think of the metadata as part of the specimen. Without it, even the best wet-lab work can't fully rescue interpretability.
Essential sample metadata
Document sample type and collection tube, draw time, processing start/finish times, centrifugation settings (g-forces, durations, temperatures), storage temperature and duration, and freeze-thaw history. If relevant, note visible hemolysis, clotting, or deviations from SOP, since these details can explain downstream anomalies.
Essential project metadata
Outline research goals, sample grouping and comparison logic, species, and whether any pilot data exist. Clear grouping logic helps bioinformatics teams pre-plan contrasts and avoid ad-hoc decisions later.
What supporting QC information is especially useful
Include concentration data (method/instrument), fragment profiles (e.g., Bioanalyzer/Fragment Analyzer traces), extraction protocol and version when submitting extracted cfDNA, and any internal QC notes. These records accelerate intake review and help teams pick appropriate library strategies within your constraints. For a neutral survey of how methylation methods interact with low-input workflows and pre-analytics, see the technology and bioinformatics overview in Frontiers in Genetics (Huang 2019) and the complementary perspective on methylation-based methods in Cancers (Galardi 2020).
Common Mistakes in cfDNA Sample Submission
Avoiding a few recurring pitfalls can save weeks and preserve interpretability.
Choosing a sample type before defining the research question
Submission strategy should support the hypothesis and comparisons. Deciding on format first can lock you into constraints that complicate the actual analysis plan.
Mixing sample formats within the same comparison set
Combining whole blood, plasma, and extracted cfDNA in one analytical comparison increases technical ambiguity. If you must mix formats across a larger project, keep each comparison internally consistent.
A simple intake triage can prevent mixed-format designs from turning into mixed-signal results: first, separate samples into format-homogeneous comparison sets (plasma-with-plasma, whole-blood-with-whole-blood); second, check whether each set has consistent handling metadata (tube type, processing window, spins, storage); third, run a small pilot QC review on the noisiest set before scaling; and finally, document any unavoidable deviations so downstream interpretation has an audit trail.
Treating extraction as a black box
Extracted cfDNA without method history or QC documentation is hard to judge. Even if the material looks fine, missing records can undermine outlier diagnostics and reduce confidence in results.
Underestimating handling variability across batches
Site-to-site and batch-to-batch drift is the most common hidden confounder in collaborative projects. Plan documentation that makes drift visible and solvable before it reaches analysis.
FAQs About cfDNA Sample Type Selection
Is plasma always better than whole blood for cfDNA methylation studies?
Not always. Plasma often provides better control of cfDNA-enriched input when separation and storage are handled consistently, but if those steps cannot be made uniform across sites, centralizing control with a whole-blood route under a single SOP can deliver more comparable inputs. The core principle is to pick the format you can execute consistently and document thoroughly.
Can I submit extracted cfDNA without detailed extraction records?
It's possible in some cases, but traceability matters. Without documentation of protocol and version, input amounts, elution conditions, fragment profile, concentration method, and storage history, it becomes harder to assess quality and to interpret downstream results. If records are limited, consider a small intake review or pilot to establish baseline behavior before scaling.
Does sample type affect downstream methylation interpretation?
Yes. Sample type influences background DNA levels, input consistency, and the confidence of downstream comparisons. Plasma prepared and stored consistently can reduce background genomic DNA; whole blood centralized under a uniform SOP can reduce site-to-site drift; extracted cfDNA is efficient when traceability is strong. In each case, documentation is what turns a format into reliable evidence.
What is the safest submission strategy for multi-site research projects?
Usually the one that is easiest to standardize and document across all samples and sites. If local separation and storage can be made uniform with training and audits, plasma is often a strong choice. If not, centralizing separation by submitting whole blood under a single SOP can keep variability in check.
What information should I prepare before requesting a quote for a cfDNA methylation project?
Have the submission format, sample number, species, expected input volume or yield, collection tube and processing timeline, centrifugation parameters, storage conditions and freeze-thaw history, extraction status and QC details if applicable, cohort structure, and analysis goal. Providing these details early makes scope and budget more stable and accelerates intake.
How to Choose the Most Practical Submission Strategy
Choose based on control, consistency, and traceability
Ask three questions: Which format lets us control pre-analytical variation the best? Which one can we apply consistently across all samples, sites, and batches? Which path creates the strongest paper trail of what happened to each sample? The answer to those questions will typically reveal the format that best fits your study.
Your project is likely ready if
You've selected a single submission format per comparison, aligned collection and processing SOPs across sites, documented timing and centrifugation parameters, planned storage and aliquoting to limit freeze-thaw, and prepared a quote-ready brief with cohort structure and analysis goals. If you want examples of scope options by assay class without changing your submission decision, see these neutral overviews: a broad human methylome panel scope and sample requirements and a method page on enzymatic methylation detection at low input. Both are background reading; your submission choice should still be driven by control, consistency, and traceability rather than the panel or chemistry.
You may need to refine the plan first if
Your comparison sets mix formats, local processing timelines vary by site or week, extraction history for existing material is thin, or storage records are incomplete. In these cases, run a short readiness check to close gaps before committing full cohorts.
Next step before requesting a quote
Organize your submission format, handling SOPs, and metadata first; then move into a targeted technical discussion or quote planning. If you want a concrete template for what providers typically need, review the intake fields and sample requirements on CD Genomics (RUO) and, when appropriate, its cfDNA methylation sequencing overview page (RUO) linked earlier. A clear brief turns complex pre-analytics into stable scope and decision-ready results.
References for further reading
- According to the evidence synthesis in NCI BEBP's Harmonizing cell-free DNA practices (Greytak 2020), tube type and processing windows materially affect cfDNA inputs.
- Comparative processing effects and double-spin benefits are discussed in Effects of collection and processing procedures on plasma ctDNA (Risberg 2018).
- Preservative and temperature impacts on cfDNA stability are detailed in The Effect of Preservative and Temperature on cfDNA Analysis (Clin Cancer Research, Hyman 2017).
- Workflow-wide risk factors are consolidated in Critical factors in circulating nucleic acids workflows (Clinical Chemistry, van der Leest 2024).
- Pre-analytical variable impacts and practical controls are summarized in Impact of preanalytical variables on cfDNA analysis (Frontiers in Cell and Developmental Biology, Peng 2024).
- Methylation technology overviews and bioinformatics considerations are presented in Cell-Free DNA Methylation Profiling-Technologies and Bioinformatics (Frontiers in Genetics, Huang 2019) and Cell-Free DNA-Methylation-Based Methods and Applications (Cancers, Galardi 2020).




