Advancements in RIP-Seq Technology: From Single-Cell Resolution to AI-Driven Insights
As the core technology to analyze the interaction between RNA binding protein (RBP) and RNA, RIP-Seq has made many important achievements in biomedical research. However, the traditional RIP-Seq technology is mainly based on population cell analysis, and it is difficult to capture the dynamic changes of cell heterogeneity and RNA-protein interaction in spatial and temporal dimensions.
In recent years, with the continuous innovation of technology and the integration of multi-omics technology, RIP-Seq has made breakthrough progress in frontier fields such as single cell resolution, spatial transcriptome research, multi-omics integration and artificial intelligence-assisted analysis, which provides a new perspective and powerful tool for deeply understanding life regulation mechanism and overcoming major diseases.
In this paper, the breakthrough progress of RIP-Seq technology in the frontier fields, as well as its application and challenges are reviewed.
RIP-Seq Technology at Single cell Resolution
Single cell RIP-Seq (scRIP-Seq) can accurately capture the interaction events between and RNA at the single cell level, break the limitation of population cell averaging data, reveal the unique patterns and dynamic differences of RBP-RNA interaction in heterogeneous cell populations, and provide a key tool for analyzing the heterogeneity of intercellular regulation in complex biological processes.
scRIP-Seq Principle
scRIP-Seq aims to break through the limitations of traditional RIP-Seq and realize accurate analysis of RNA-protein interaction in a single cell. Its technical process is optimized on the basis of traditional RIP-Seq.
- First, single cell is separated by single cell separation technology, such as microfluidic technology and fluorescence activated cell sorting (FACS).
- Then, RNA immunoprecipitation was carried out for each single cell, and the RNA-protein complex bound to the target RBP was captured by using specific antibodies. Because the content of RNA and protein in single cell is extremely low, it is necessary to amplify the precipitated RNA efficiently to meet the demand of high-throughput sequencing. At present, the commonly used RNA amplification methods include linear amplification technology based on reverse transcription and PCR to minimize amplification deviation and ensure the accuracy of data.
- Finally, the amplified RNA was high-throughput sequencing, and the binding spectrum of RBP at single cell level was analyzed by bioinformatics analysis.
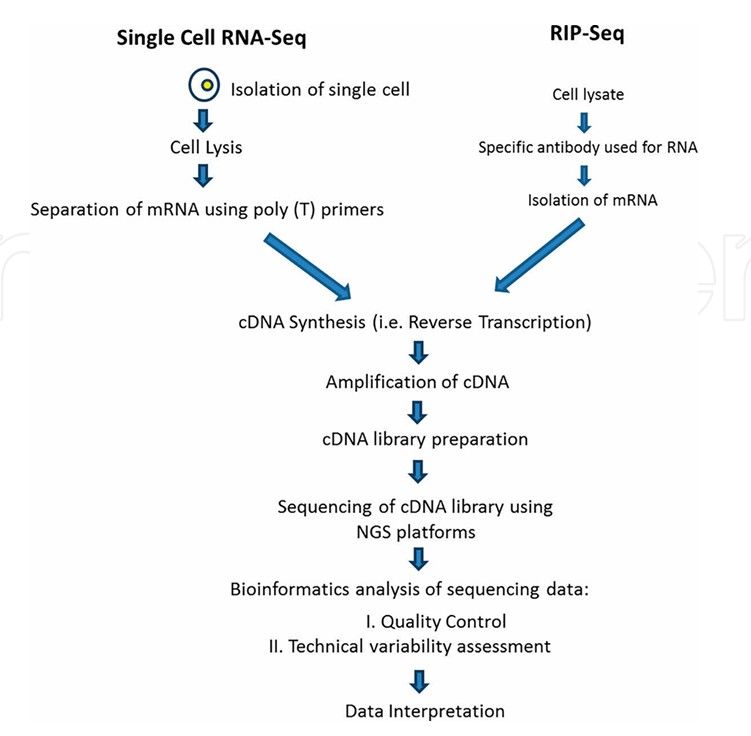 Flowchart of scRNA-seq and RIP-Seq (Nakul et al., 2022)
Flowchart of scRNA-seq and RIP-Seq (Nakul et al., 2022)
Practical Application and Challenges
scRIP-Seq shows key value in analyzing the regulation of cell heterogeneity. It can accurately reveal the interaction mode between RNA and protein in a single cell by sorting single cells through microfluidic, specifically capturing RNA-protein complex and combining with efficient amplification technology.
- Cancer Research: Identifies RBPs driving distinct tumor cell subpopulations, uncovering subtype-specific therapeutic targets.
- Embryonic Development: Tracks dynamic RBP-RNA interactions during cell differentiation, clarifying regulatory roles in cell fate decisions.
- Low RNA/protein content in single cells demands ultra-sensitive protocols to avoid masking low-abundance interactions.
- Data complexity requires advanced bioinformatics to decode high-dimensional, sparse datasets and identify meaningful heterogeneity.
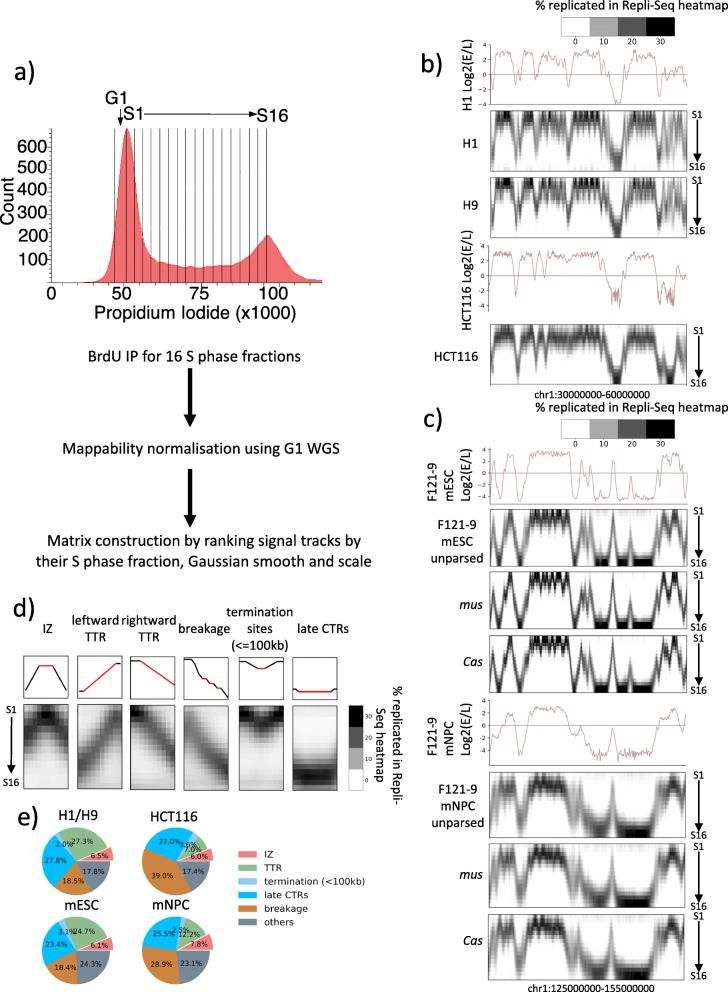 High-resolution Repli-Seq produces robust and reproducible heatmaps that annotate features of replication at fine temporal resolution (Zhao et al., 2020)
High-resolution Repli-Seq produces robust and reproducible heatmaps that annotate features of replication at fine temporal resolution (Zhao et al., 2020)
Service you may intersted in
Learn More:
RIP-Seq Combined with Spatial Transcriptome
The combination of spatial transcriptome and RIP-Seq provides a revolutionary tool for analyzing the spatio-temporal heterogeneity of RNA-protein interaction in biological samples. The combination of the two can accurately locate the regional specificity of RBP regulatory activity on the premise of preserving the spatial background of tissues.
Principles and Methods
Spatial transcriptome technology can preserve the spatial location information of cells in situ and detect gene expression at the same time. The combination of spatial transcriptome and RIP-Seq aims to combine their advantages and realize the analysis of RNA-protein interaction in spatial dimension. The specific method:
- Process tissue samples by using spatial transcriptome technology to obtain the spatial information of gene expression.
- Then, RIP-Seq experiment was carried out on the same tissue sample to identify the binding relationship between RNA and protein.
- Finally, through bioinformatics integration analysis, the binding spectrum of RBP is combined with the spatial distribution of gene expression, thus revealing the spatial specificity of RNA-protein interaction in tissue microenvironment.
Application and Significance
In tumor research, tumor microenvironment plays a key role in the occurrence, development and metastasis of tumors. The combination of spatial transcriptome and RIP-Seq can be used to analyze the difference of RNA-protein interaction in different regions of tumor tissue.
In the nervous system, different brain regions have unique functions, and their gene expression and regulation mechanisms are significantly different. This combined technique can be used to study the changes of RNA-protein interaction in specific brain regions in nervous system diseases.
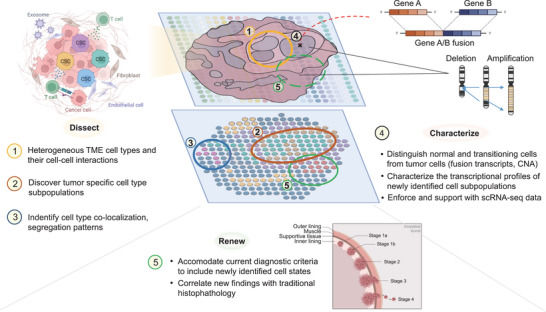 Applications of spatial transcriptomics in cancer research (Park et al., 2023)
Applications of spatial transcriptomics in cancer research (Park et al., 2023)
Localization of RNA-protein Interaction in Situ
In addition to the combination of spatial transcriptome and RIP-Seq, in-situ RNA-protein interaction localization technology is also developing continuously. Based on the improvement of fluorescence in situ hybridization (FISH) and immunofluorescence (IF) technology, RNA-protein interaction can be directly observed in situ in cells and tissues. By designing specific probes and antibodies, these techniques label RNA and RBP respectively, and use the co-location of fluorescent signals to indicate the binding of RNA-protein.
In-situ RNA-protein interaction mapping technology can visually present the spatial distribution of RNA-protein interaction, which complements RIP-Seq data and provides more comprehensive information for further understanding the biological function of RNA-protein interaction.
Multiomics Integration Analysis with RIP-Seq
The integrated analysis of RIP-Seq and multi-omics technology is becoming the core strategy to analyze the complex biological mechanism, which can construct a multi-dimensional correlation map from RNA-protein interaction to gene expression regulation and metabolic pathway network, and systematically reveal the synergistic law of multi-level molecular events in the process of disease occurrence or development.
Combined CLIP-Seq, RNA-Seq and Proteomics Data
CLIP-Seq can accurately identify the direct binding site between RBP and RNA at the whole transcriptome level. RNA-Seq can be used to detect the expression level of genes. Protein genomics can analyze the expression and modification of protein. By combining RIP-Seq with CLIP-Seq, RNA-Seq and protein data, the regulatory network of RNA-protein interaction can be comprehensively analyzed from multiple levels.
- RIP-Seq provides the binding information of RBP and RNA.
- CLIP-Seq further defines the binding site.
- RNA-Seq reflects the change of gene expression, and proteomics data reveals the regulation of protein level.
Through the integrated analysis of bioinformatics, the association network between RNA-protein interaction, gene expression and protein regulation can be constructed.
In the study of cardiovascular diseases, researchers combined RIP-Seq, CLIP-Seq, RNA-Seq and proteomics data to find a RBP regulatory network related to myocardial cell function. The binding targets of the RBP were identified by RIP-Seq and CLIP-Seq. RNA-Seq analysis showed that the expression of genes related to these targets changed in the state of cardiovascular disease, and proteomics data revealed the influence of the RBP on the expression and modification of downstream protein. The results of integration analysis show that RBP participates in the contraction and metabolism of myocardial cells by regulating the expression of a series of genes and protein function, and its dysfunction is closely related to the occurrence and development of cardiovascular diseases.
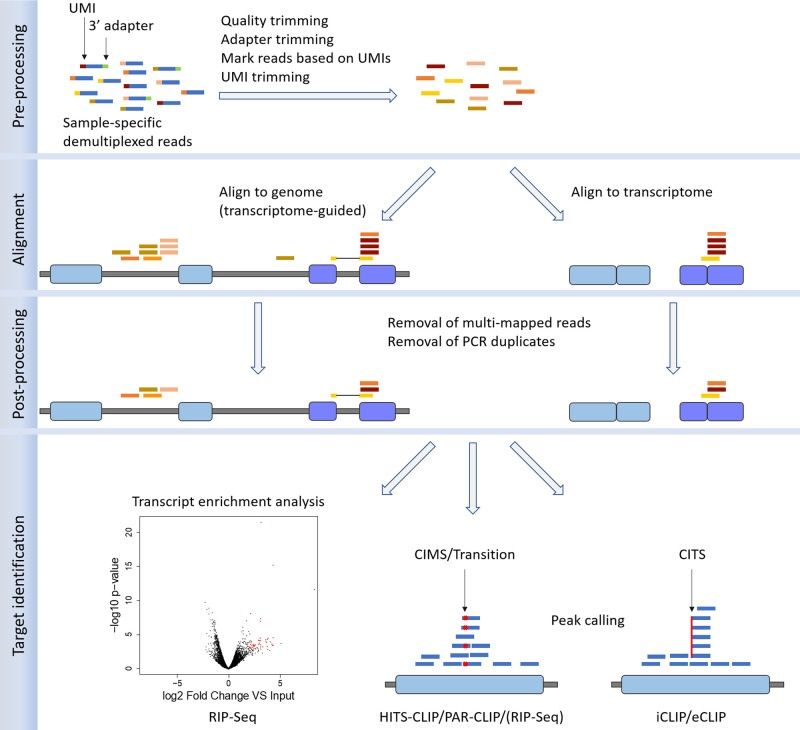 Bioinformatics analysis of CLIP-Seq and RIP-Seq data (Colantoni et al., 2020)
Bioinformatics analysis of CLIP-Seq and RIP-Seq data (Colantoni et al., 2020)
Advantages and Challenges of Integrated Analysis
Multiomics integration analysis can provide more comprehensive and systematic biological information, which is helpful to deeply understand the complex life regulation mechanism.
- By integrating different omics data, we can verify and supplement the research results from many angles and improve the reliability of the research conclusions.
- In addition, the integration of omics can also find new regulatory pathways and biomarkers, providing more potential targets for the diagnosis, treatment and prognosis evaluation of diseases.
- Multiomics data have different characteristics and formats, and the standardization and normalization of data is the primary problem faced by integrated analysis.
- Multiomics experiment has high cost and complex technical requirements. How to optimize the experimental design and process and improve the data quality and efficiency is also an important aspect to be considered.
Application of Artificial Intelligence in RIP-Seq
In RIP-Seq research, artificial intelligence is showing great potential. Through deep learning algorithm, AI can efficiently analyze the massive data generated by RIP-Seq and accurately identify the interaction pattern between RNA binding protein (RBP) and RNA.
Prediction of RBP Binding Sites by Deep Learning
Deep learning has strong ability of feature extraction and pattern recognition, which shows great potential in RIP-Seq data analysis. Researchers use deep learning algorithms, such as convolutional neural network (CNN) and recurrent neural network (RNN), to analyze RIP-Seq data to predict the binding sites of RBP.
- Firstly, RIP-Seq data is converted into a format suitable for the input of deep learning model, such as sequence feature matrix.
- Then, by training the deep learning model, the sequence characteristics and structural characteristics of RBP binding sites are learned.
- The experimental results show that the deep learning model has high accuracy and sensitivity in predicting RBP binding sites, and can find potential binding sites that are difficult to identify by traditional methods.
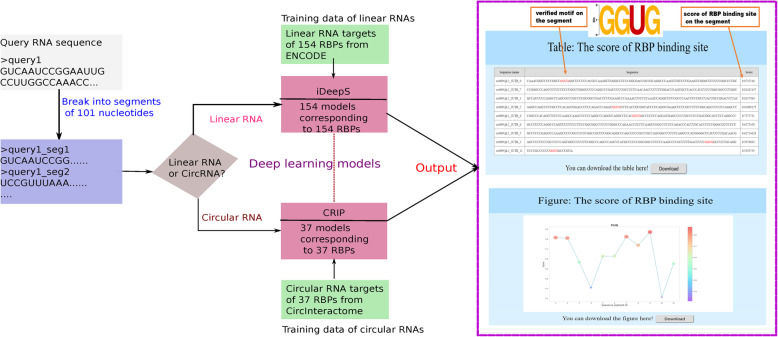 The workflow of RBPsuite webserver (Pan et al., 2020)
The workflow of RBPsuite webserver (Pan et al., 2020)
Artificial Intelligence Assisted Data Analysis and Mining
In addition to predicting RBP binding sites, artificial intelligence can also be used in other analysis links of RIP-Seq data.
- Using machine learning algorithm to cluster RIP-Seq data can automatically identify cell subsets or sample categories with similar RNA-protein interaction patterns.
- Used for data visualization and interpretation, which can help researchers better understand complex RIP-Seq data by constructing intuitive graphs and models.
- Artificial intelligence also plays an important role in data mining, which can mine potential biological laws and regulatory mechanisms from a large number of RIP-Seq data.
New Sequencing Technologies Based on RIP-Seq
RIP-Seq, as the core technology for analyzing RNA-protein interaction, is constantly deriving new technical branches, which opens up a new path for revealing the dynamic regulation network of RNA in complex biological samples and promotes the precise transformation of basic research and clinical application.
DO-RIP-Seq
DO-RIP-Seq is a new RIP-Seq derivative technology, which is mainly used to detect RNA related to chromatin. Its technical principle is to enrich RNA that combines with RBP and interacts with chromatin in the process of RNA immunoprecipitation. Through DO-RIP-Seq, researchers can deeply understand the regulation of RNA at chromatin level, such as RNA-mediated chromatin modification and gene transcription regulation.
In the study of gene expression regulation, DO-RIP-Seq has been preliminarily applied to analyze the interaction between non-coding RNA and chromatin, and it is found that some long-chain non-coding RNA can affect the structure and function of chromatin by binding with specific proteins on chromatin, thus regulating gene expression. With the continuous improvement of technology, DO-RIP-Seq is expected to play a greater role in the field of epigenetics and provide a powerful tool for revealing new mechanisms of gene expression regulation.
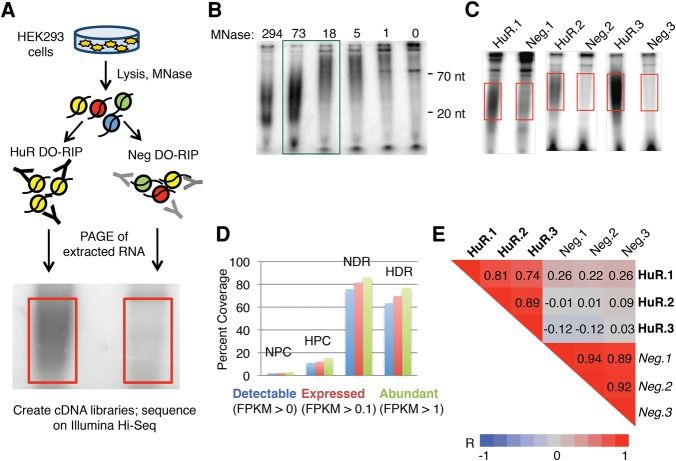 A schematic representation of DO-RIP-seq procedure (Nicholson et al., 2017)
A schematic representation of DO-RIP-seq procedure (Nicholson et al., 2017)
Other Potential Emerging Technologies
Besides DO-RIP-Seq, RIP-Seq technology has many potential development directions.
- Combined with CRISPR technology, a tool that can accurately regulate the interaction between specific RBP and RNA is developed to study the function of RNA-protein interaction in specific biological processes.
- The improvement of RIP-Seq technology based on nanotechnology is expected to further improve the sensitivity and resolution of detection and realize the accurate analysis of RNA-protein interaction in micro samples.
With the continuous innovation of technology and the strengthening of interdisciplinary cooperation, more multifunctional and powerful RIP-Seq emerging derivative technologies will emerge in the future, which will promote the development of life science research to a higher level.
Conclusion
RIP-Seq technology has shown inestimable value from analyzing RNA-protein interaction network, revealing the mysterious function of non-coding RNA, and helping to explore disease mechanism and develop precision medicine. RIP-Seq technology has made remarkable progress in frontier fields such as single cell resolution, spatial transcriptome combination, multi-omics integration, artificial intelligence application and emerging derivative technologies, which has brought new opportunities and breakthroughs for life science research.
However, these cutting-edge technologies also face many challenges in the development process, such as the complexity of technology, the difficulty of data analysis and the cost of experiments. In the future, it is necessary to further strengthen technological innovation and interdisciplinary cooperation, constantly optimize experimental methods and data analysis algorithms, overcome technical bottlenecks, give full play to the potential of RIP-Seq cutting-edge technology, and provide stronger technical support for deeply understanding life regulation mechanism and overcoming major diseases.
References
- Nakul D M, Priya S K., et al. "Gene Expression and Transcriptome Sequencing: Basics, Analysis, Advances." Gene Expression. 2022 8: 14 http://dx.doi.org/10.5772/intechopen.105929
- Zhao PA, Sasaki T, Gilbert DM. "High-resolution Repli-Seq defines the temporal choreography of initiation, elongation and termination of replication in mammalian cells." Genome Biol. 2020 21(1): 76 https://doi.org/10.1186/s13059-020-01983-8
- Park HE, Jo SH., et al. "Spatial Transcriptomics: Technical Aspects of Recent Developments and Their Applications in Neuroscience and Cancer Research." Adv Sci (Weinh). 2023 10(16) :e2206939 https://doi.org/10.1002/advs.202206939
- Colantoni A, Rupert J., et al. "Zooming in on protein-RNA interactions: a multi-level workflow to identify interaction partners." Biochem Soc Trans. 2020 48(4): 1529-1543 https://doi.org/10.1042/bst20191059
- Pan X, Fang Y., et al. "RBPsuite: RNA-protein binding sites prediction suite based on deep learning." BMC Genomics. 2020 21(1): 884 https://doi.org/10.1186/s12864-020-07291-6
- Nicholson CO, Friedersdorf M, Keene JD. "Quantifying RNA binding sites transcriptome-wide using DO-RIP-seq." RNA. 2017 23(1): 32-46 https://doi.org/10.1261/rna.058115.116




