TAIL Iso-Seq: Poly(A) Tail Profiling & APA Mapping Solutions
CD Genomics offers expert TAIL Iso-Seq services for high-resolution analysis of mRNA poly(A) tail length and transcriptome complexity. Powered by Nanopore long-read sequencing, our platform delivers full-length transcript information with accurate polyadenylation site mapping. Ideal for exploring alternative polyadenylation, RNA stability, and gene regulation across species or developmental stages. Unlock new insights in poly(A) tail dynamics with our end-to-end solution for poly(A) tail sequencing.

- Key Technical Advantages of TAIL Iso-Seq
- Key Applications of TAIL Iso-Seq
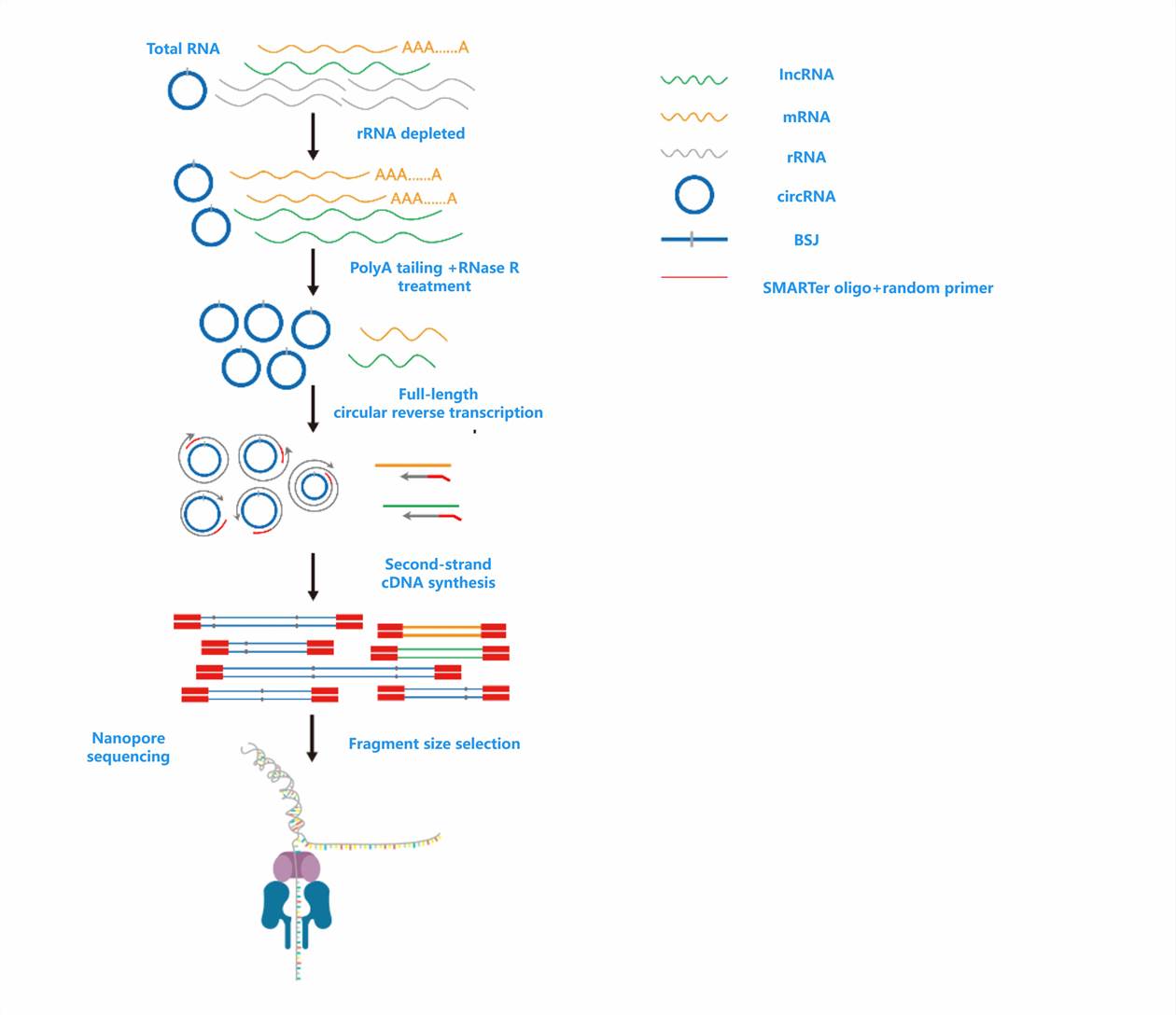
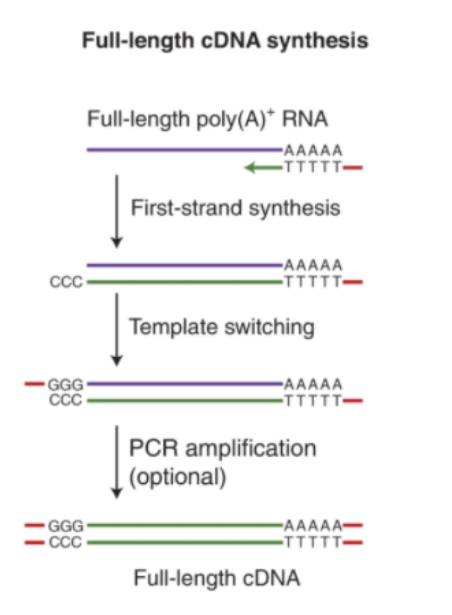
- Library Preparation Workflow
- Bioinformatics
- Deliverables
- Sample Requirements
- Other Sequencing Options for Poly(A) Analysis by CD Genomics
- FAQ – Your TAIL Iso-Seq Questions Answered
- Case Studies
Key Technical Advantages of TAIL Iso-Seq
Full-length transcript accuracy & isoform discovery
Nanopore long-read sequencing captures complete mRNA molecules with their native poly(A) tails—avoiding assembly errors common in short-read RNA-seq and enabling high-confidence identification of splice variants, fusion genes, and novel isoforms.
Single-molecule resolution of poly(A) tails
TAIL Iso-Seq directly measures the length and composition of poly(A) tails on each read. This enables precise poly(A) length profiling without PCR bias, using tools like nanopolish, tailfindr, or BoostNano.
Comprehensive polyadenylation site and APA analysis
By mapping full-length reads, TAIL Iso-Seq reveals exact polyadenylation cleavage sites and enables alternative polyadenylation (APA) profiling—key to understanding differential 3′UTR usage and transcript regulation.
Quantitative poly(A) tail length comparisons
Nanopore-based protocols—including Nano3P-seq and TERA-Seq—support high-resolution comparisons of tail length distributions across conditions or groups, using statistical tools like NanopLen for differential tail dynamics analysis.
Versatile detection of poly(A) and non-A tail variants
Protocols such as Nano3P-seq detect non-adenosine tail additions (e.g., uridylation) and profile poly(A) tails on diverse RNA types (mRNA, lncRNA, mitochondrial rRNA).
High sensitivity—even for low-abundance transcripts
Long-read sequencing covers rare or low-expression isoforms without needing complex amplification, offering deep transcriptome insight even with limited starting material
Key Applications of TAIL Iso-Seq
Alternative Polyadenylation (APA) Analysis
- Precisely map poly(A) cleavage sites and 3′ UTR diversity across transcripts.
- APA patterns often correlate with tissue types and developmental stages, impacting gene regulation. Steady-state analyses in plants showed APA-linked tail length variation across tissues
Accurate Poly(A) Tail-Length Profiling
- Obtain per-isoform tail length distributions without PCR bias or assembly errors.
- Mammalian embryo studies using Nano3P-seq revealed isoform-specific tail dynamics linked to mRNA stability.
Transcript Isoform Discovery
- Detect splice variants, novel isoforms, and fusion transcripts using full-length reads.
- Ideal for complex transcriptomes, including non-model organisms.
mRNA Stability & Translation Efficiency Studies
- Tail length is a known determinant of mRNA half-life and translation potential.
- Plant data showed long-tailed transcripts often have short half-lives, while stable ones carry shorter tails.
Broad Transcriptome Coverage
- Captures coding and non-coding RNAs—lncRNA, mitochondrial rRNAs, and more.
- Nano3P-seq demonstrated poly(A) detection even in 16S mitochondrial rRNA from zebrafish and mouse.
Cross-Species & Tissue Comparison
- Suitable for comparative studies across species or developmental stages; plant atlas recorded >120 million poly(A) profiles across multiple species.
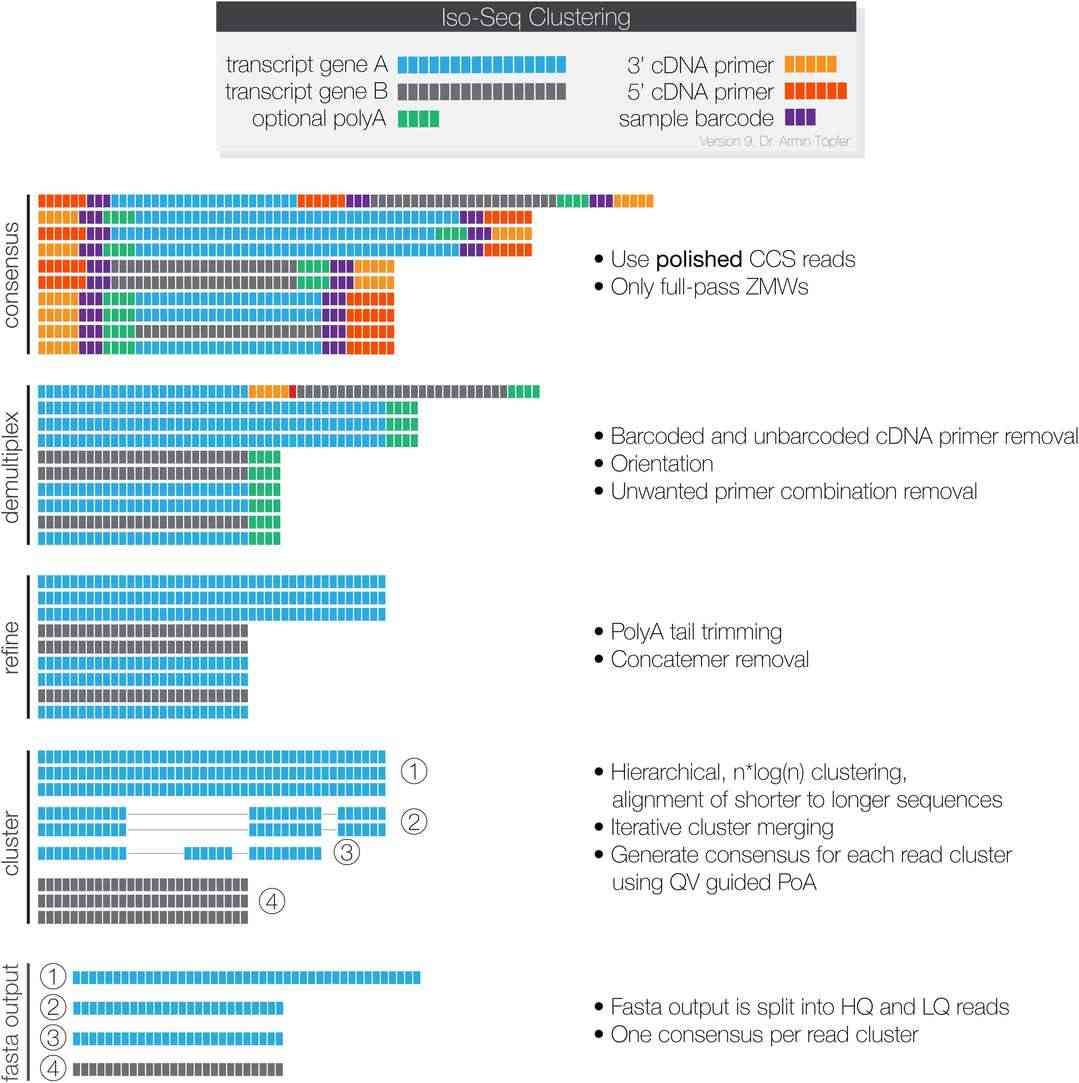
Library Preparation Workflow

Bioinformatics
Our end-to-end TAIL Iso-Seq service includes comprehensive processing and analysis of Nanopore long-read data. Clients receive the following outputs:
- Raw sequencing files
- .fast5 (raw signal) + .fastq (basecalled reads) to support downstream reanalysis or custom pipelines.
- Full-length transcript assemblies
- High-quality, full-length isoform sequences, annotated with transcript IDs, splice variants, and fusion transcripts.
- Poly(A) tail length metrics
- Per-read and per-isoform poly(A) length distributions. Native signal-based measurement ensures single-molecule resolution. Software such as nanopolish or tailfindr are used.
- Polyadenylation site & APA mapping
- Precise mapping of poly(A) cleavage sites and global assessment of alternative polyadenylation (APA) across samples.
- Differential poly(A) tail analysis
- Statistical comparison of poly(A) length distributions between groups/conditions. We apply robust models (e.g., linear mixed-effects) to identify significant tail-length shifts .
- Expression quantification & correlation
- Transcript-level expression estimates (TPM counts), integrated with poly(A) tail data to explore tail–expression relationships.

Deliverables
- User-friendly deliverables include:
- Tail length distribution plots (histograms, violin plots)
- Heatmaps of tail length vs. expression
- APA site usage diagrams
- Interactive summary report (PDF/HTML)
Sample Requirements
| Sample Type | Minimum Input Recommended |
| Animal cells | ≥ 1×106 cells |
| Animal tissue | ≥ 1 g |
| Plant tissue | ≥ 3 g |
| Eukaryotic microbes | ≥ 300 mg wet weight or ≥ 1×106 cells |
| Total RNA | ≥ 1 µg (RIN ≥ 7 recommended) |
Other Sequencing Options for Poly(A) Analysis by CD Genomics
In addition to our flagship TAIL Iso-Seq, CD Genomics offers several complementary sequencing services tailored to explore poly(A) tails depending on research goals, sample type, and downstream applications:
- Poly(A)-Seq (Illumina + Oligo(dT) Enrichment)
- Transcriptome-wide mapping of polyadenylation sites and tail lengths using high-throughput short-read sequencing.
- Ideal for comprehensive profiling of mRNA stability, 3′UTR usage, and correlation with gene expression).
- Nanopore Direct RNA-Seq
- PCR-free, single-molecule sequencing of native RNA with intact poly(A) tails and base modifications.
- Enables direct poly(A) tail length estimation and detection of RNA modifications in the same reads.
- Standard mRNA-Seq (Illumina)
- Enriches polyadenylated mRNAs for broad gene expression analysis.
- Supports bulk quantification of tail-regulated genes and library prep via oligo(dT) capture.
Choosing the Right Option
| Service | Best For |
| TAIL Iso-Seq | Isoform-level tail profiling, APA mapping, novel transcript discovery |
| Poly(A)-Seq | High-throughput tail and 3′UTR site profiling across many samples |
| Direct RNA-Seq | Native RNA with poly(A) tail + modification detection in individual reads |
| Standard mRNA-Seq | Cost-effective gene expression studies in poly(A)+ transcriptomes |
Feel free to request a custom poly(A) analysis package combining multiple platforms—for example, pairing TAIL Iso-Seq with Direct RNA-Seq to capture both structural and epitranscriptomic features.
FAQ – Your TAIL Iso-Seq Questions Answered
Q1: Can you work with low-input or precious clinical samples?
Yes. We offer optimized protocols and consultation for samples with limited RNA yield, even in clinical or rare-sample workflows.
Q2: Do you support multi-condition or longitudinal study designs?
Absolutely. Our analysis pipeline includes normalization and statistical comparison of poly(A) tail length across experimental groups or time points.
Q3: Can you integrate poly(A) profiling data with existing RNA-seq or proteomics?
Yes. Reports can include cross-platform correlation—e.g., linking tail length changes to expression or protein abundance.
Q4: Are processed data and intermediate files available for follow-up analysis?
Clients receive full bioinformatics outputs (e.g., fastq, assembled transcripts, tail length tables) and visualizations. Raw signal files may be provided upon request.
Q5: Do you provide customizable data formats or integrations?
Yes. We support custom outputs including differential analysis tables, interactive plots, or formatted results ready for LIMS or downstream pipelines.
Q6: Is guidance available for experimental design and data interpretation?
Our scientific team offers consultation at no extra cost—helping you plan sample prep, understand results, and align with publication goals.
Q7: How do you mitigate bias in poly(A) tail length estimation?
We sequence native RNA or minimally-amplified cDNA to avoid PCR-induced distortions. For direct RNA-seq, we retain .fast5 files to run tools like nanopolish or tailfindr for accurate tail length estimation .
Q8: Can you detect and quantify non-A residues or tail heterogeneity?
Yes. By analyzing raw signal data—especially with protocols like Nano3P-seq or SM-PATseq—we detect uridylation, guanylation and mixed-tail patterns at single-nucleotide resolution .
Q9: How do you handle systematic errors in Nanopore direct RNA sequencing for homopolymer tails?
We apply rigorous basecalling with optimized models (e.g., RNA accuracy ≥90%), followed by signal-level correction and tail trimming tools to correct homopolymer-induced deletions .

Case Studies
Nano3P-seq: Developmental RNA Tail Dynamics – Nature Methods (2023)
https://doi.org/10.1038/s41592-022-01714-w
Nano3P-seq uses Nanopore cDNA sequencing with a 3′ end capture method to quantify RNA abundance, tail length, and tail composition at single-read resolution—without PCR bias. Applied to mouse and zebrafish embryos, the study detected dynamic, isoform-specific poly(A) tail changes that correlate with mRNA decay, and even identified polyadenylated 16S mitochondrial rRNAs. This work establishes a robust, high-resolution approach for transcriptome-wide poly(A) profiling.

For research purposes only, not intended for personal diagnosis, clinical testing, or health assessment