How to Analyze Iso-Seq Data
At a glance:
- Introduction
- Advantages of Long-Read Sequencing Technology
- Unique Value of Iso-Seq in Tumor Transcriptomics and Variant Analysis
- Comparison with Short-Read Transcriptome Data
- Analyzing Iso-Seq Data
- Conclusion
Iso-Seq, a long-read sequencing technology developed by Pacific Biosciences (PacBio), enables direct full-length RNA sequencing, offering a high-resolution view of transcriptomes. Unlike short-read RNA-seq, Iso-Seq captures complete transcripts from the 5' cap to the poly-A tail, facilitating accurate analysis of alternative splicing, transcription start/end sites, and polyadenylation events. This eliminates the need for computational assembly and improves isoform detection, especially in complex tissues and cancer. Iso-Seq excels in identifying rare isoforms, fusion genes, and structural variants with tools like IsoSeq3, DeBreak, and FusionSeeker. It supports precise transcript annotation, fusion breakpoint mapping, and SV characterization at single-molecule resolution. Despite higher costs and lower throughput, Iso-Seq's ability to resolve transcriptomic complexity makes it invaluable in cancer, neuroscience, and developmental biology. Its integration into standard workflows is expanding, offering researchers a powerful means to investigate transcript structure and regulatory dynamics across diverse biological contexts.
Introduction
In the rapidly evolving field of genomics, the ability to accurately capture and analyze the full complexity of the transcriptome is paramount. Iso-Seq, a long-read sequencing technology developed by PacBio offers a transformative approach to transcriptome analysis. Unlike traditional short-read RNA sequencing, which often requires computational assembly of fragmented transcripts, Iso-Seq directly sequences entire RNA molecules from the 5' cap to the poly-A tail. This full-length resolution provides researchers with an unprecedented view into transcript structure, alternative splicing events, isoform diversity, and regulatory mechanisms.
Comprehensive transcriptome profiling is crucial for understanding the dynamic nature of gene expression, especially in tissues with high isoform complexity or during disease progression. Conventional RNA-seq approaches using short reads often miss low-abundance isoforms, complex splice junctions, or non-canonical transcripts, leading to incomplete or fragmented insights. In contrast, Iso-Seq delivers a more holistic, high-confidence representation of the transcriptome, significantly improving our ability to study gene function, tissue-specific expression, and disease-related transcriptomic alterations.
The utility of Iso-Seq extends far beyond basic biology. It has emerged as a powerful tool in biomedical research, especially in cancer biology, where transcriptional heterogeneity, fusion genes, and structural variations often drive pathogenesis. By offering single-molecule resolution and avoiding assembly bias, Iso-Seq allows researchers to capture subtle but critical transcriptomic variations that are often invisible to short-read approaches.
Advantages of Long-Read Sequencing Technology
A defining strength of the Iso-Seq platform is its capability to sequence entire RNA molecules without interruption, capturing transcripts in their full-length form.
Enhanced Detection of Splicing Variants:
Iso-Seq facilitates the direct identification of transcript variants generated through alternative splicing mechanisms such as exon skipping, use of non-canonical 5' or 3' splice sites, intron retention, and mutually exclusive exons. These splicing patterns are crucial for regulating gene expression and cellular specificity. Aberrations in these processes are frequently associated with various pathologies, especially in oncological and neurological conditions.
High-Resolution Mapping of Transcription Start and Termination Sites:
With complete transcript coverage, Iso-Seq enables precise annotation of both transcription initiation (TSS) and termination sites (TES). This detailed mapping supports investigations into promoter dynamics and polyadenylation site utilization, both of which influence transcript lifespan, cellular localization, and translational efficiency.
Accurate Profiling of 3' UTRs and Polyadenylation Events:
Iso-Seq data provides refined insights into alternative polyadenylation (APA), a widespread regulatory mechanism affecting transcript stability and function. APA plays significant roles in cell-type identity and is often exploited in cancer to evade miRNA targeting or to alter mRNA degradation pathways.
Assembly-Free Isoform Reconstruction:
In contrast to short-read sequencing, which typically relies on computational reconstruction of transcript structures:
Minimized Misassembly Artifacts: By removing the need for in silico transcript assembly, Iso-Seq significantly reduces erroneous isoform reconstruction caused by read ambiguity or repetitive genomic regions.
Streamlined Computational Workflow: The clarity of full-length reads simplifies downstream bioinformatics, enabling faster data interpretation and more intuitive analyses.
Reliable Isoform Quantification: Iso-Seq offers unambiguous discrimination between closely related isoforms and gene family members, ensuring more accurate quantification-especially critical in the context of paralogous genes.
Unique Value of Iso-Seq in Tumor Transcriptomics and Variant Analysis
Iso-Seq has become an indispensable tool in cancer research, where transcriptomic complexity is often heightened:
Detection of Cancer-Specific Isoforms: Tumors frequently express unique transcript isoforms due to somatic mutations or epigenetic reprogramming. Iso-Seq can detect and characterize these aberrant isoforms, some of which may function as neoantigens or therapeutic targets.
Discovery of Novel Fusion Genes: Gene fusions are hallmarks of many cancers (e.g., BCR-ABL in leukemia, TMPRSS2-ERG in prostate cancer). Iso-Seq's long reads enable precise identification of fusion breakpoints, fusion partners, and retained introns or alternative exons in fusion transcripts-information that is often lost in short-read data.
Characterization of Complex Structural Variants (SVs): Structural rearrangements like inversions, duplications, and translocations can have profound effects on gene expression and splicing. Iso-Seq allows researchers to detect these SVs at the transcript level, directly linking genomic structure to functional transcript output.
Resolution of Tumor Heterogeneity: The single-molecule resolution of Iso-Seq captures rare transcripts and heterogeneous subpopulations within a tumor, providing insights into clonal evolution, drug resistance, and metastatic potential.
Comparison with Short-Read Transcriptome Data
The following table provides a systematic comparison between Iso-Seq and short-read RNA-Seq technologies, highlighting their distinct characteristics across key performance metrics:
| Feature | Iso-Seq | RNA-Seq (Short-Read) |
| Read Length | ~10–15 kb (HiFi reads) | ~150–300 bp |
| Transcript Coverage | Full-length | Fragmented |
| Isoform Resolution | Direct, accurate | Indirect, assembly-dependent |
| Splice Junction Accuracy | High | Lower, inference-based |
| PolyA and TSS Detection | Direct | Indirect |
| Structural Variant & Fusion Detection | High-resolution | Limited |
| Throughput | Lower | Very high |
| Cost per Base | Higher | Lower |
Analyzing Iso-Seq Data
Part 1: Basic Processing Workflow with IsoSeq3
IsoSeq3 is the official analysis pipeline provided by PacBio. Below is a step-by-step overview:
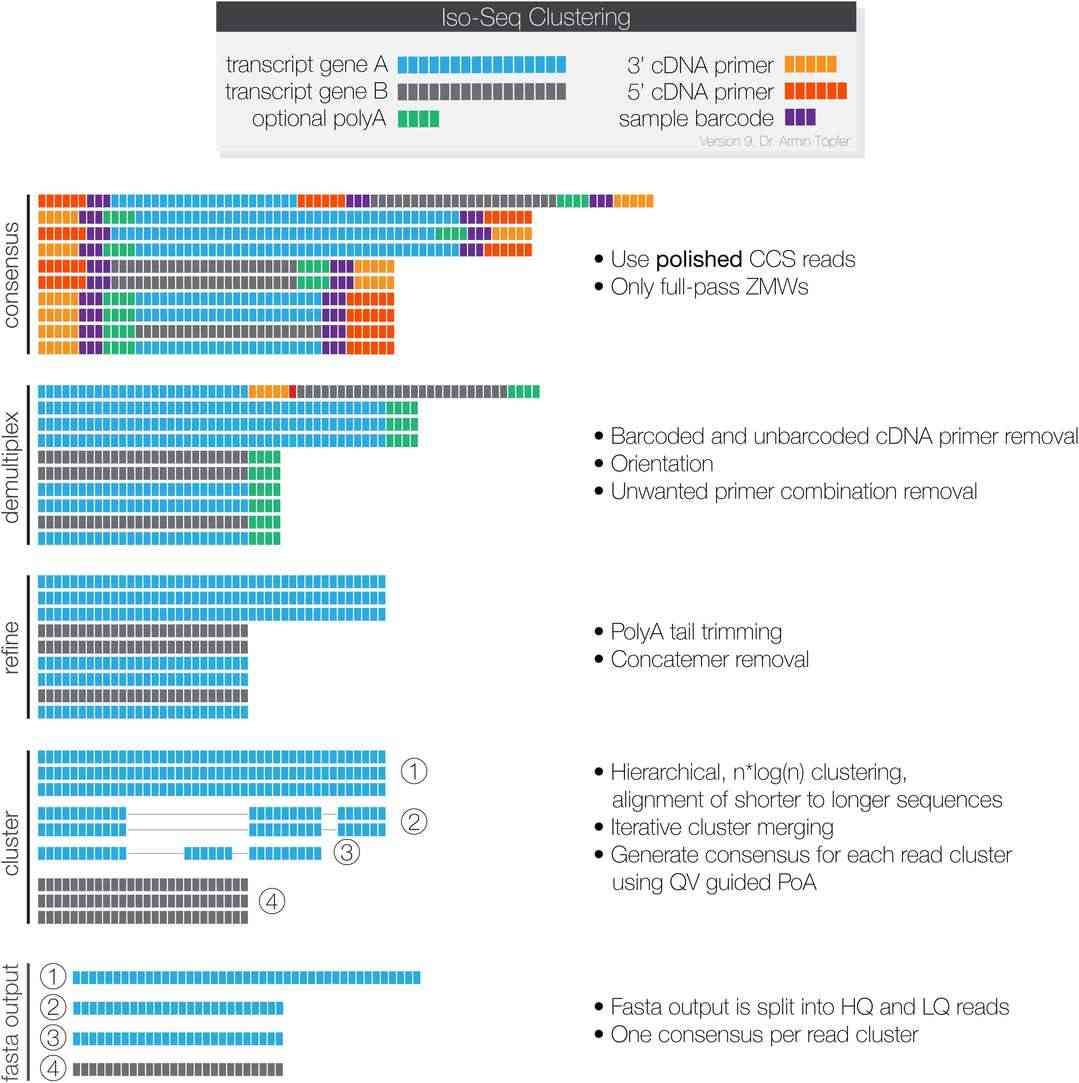 Figur1 . Workflow of IsoSeq3.(Ramberg S. et al, 2021)
Figur1 . Workflow of IsoSeq3.(Ramberg S. et al, 2021)
Install IsoSeq3
#bash conda install isoseq3
Generate Circular Consensus Sequences (CCS)
Convert subreads into high-quality CCS reads using multiple passes of the polymerase:
#bash ccs movie.subreads.bam movie.ccs.bam --min-rq 0.9
Remove Primers and Classify Full-Length Reads
Trim adapter/primer sequences and identify full-length non-chimeric reads:
#bash lima movie.ccs.bam primers.fasta movie.fl.bam --isoseq --peek-guess
Refine FLNC Reads and Trim Poly-A Tails
Remove poly-A tails and confirm 5' and 3' completeness:
#bash isoseq3 refine movie.fl.bam primers.fasta movie.flnc.bam
Cluster and Polish Transcripts
Group similar reads to create high-quality consensus isoforms:
#bash isoseq3 cluster movie.flnc.bam movie.polished.bam -verbose
Align Consensus Reads to Reference Genome
Use pbmm2 for accurate spliced alignment:#bash pbmm2 align reference.fasta movie.polished.bam movie.aligned.bam --preset ISOSEQ –sort
Collapse Redundant Transcripts and Annotate
Merge redundant isoforms and output gene/transcript models:
#bash isoseq3 collapse movie.aligned.bam collapsed.gff
Follow-up analysis includes:
Annotation comparison with GENCODE/RefSeq using SQANTI3
Isoform-level expression quantification
Functional enrichment analysis (GO, KEGG)
Part 2: Identifying Structural Variants with DeBreak
DeBreak is a long-read-aware SV detection tool designed for high accuracy and transcript-aware breakpoint mapping. DeBreak is especially valuable when integrated with Iso-Seq for capturing transcript-level SV consequences, such as fusion transcripts with disrupted domains or SV-driven exon skipping.
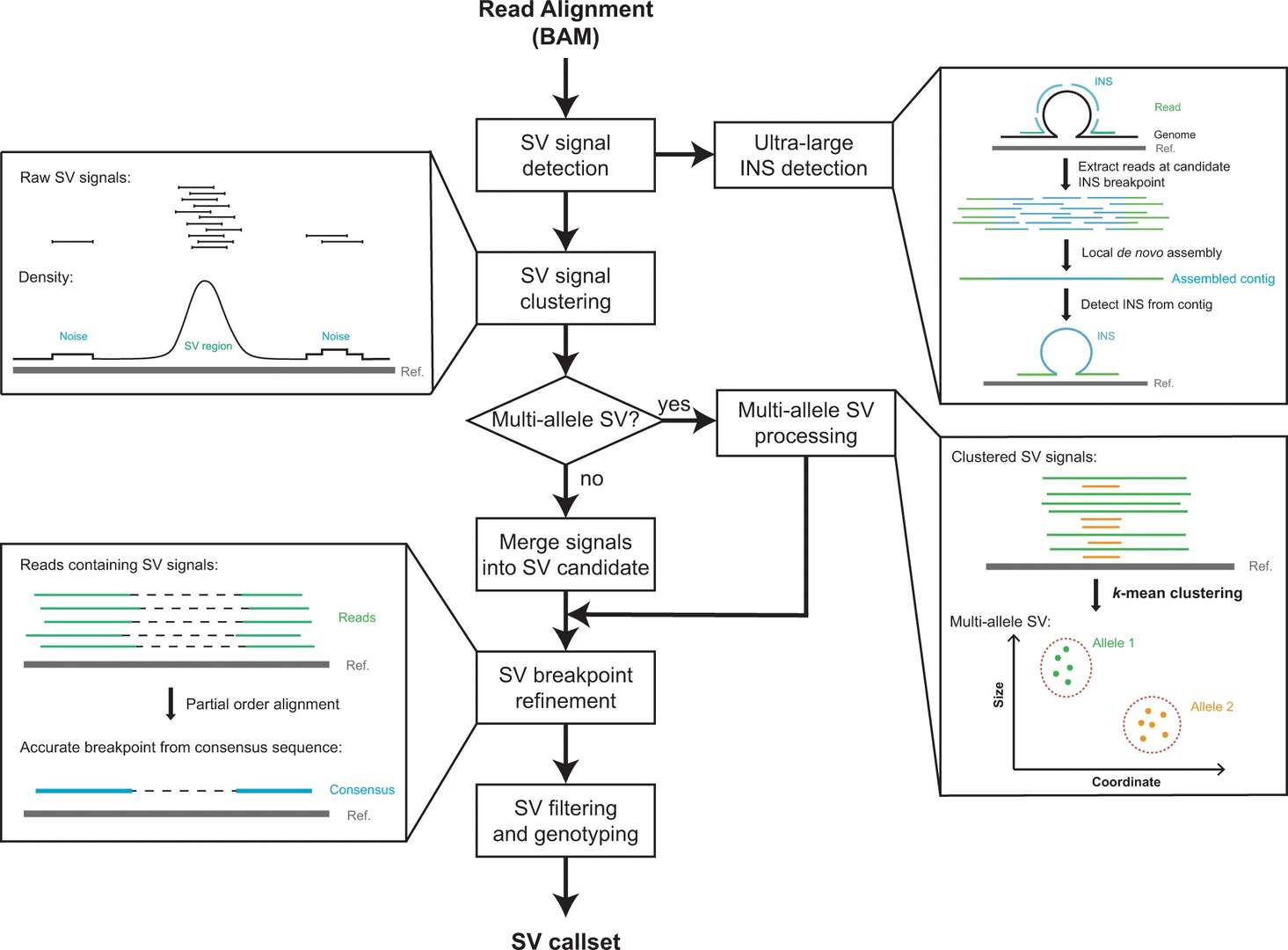 Figur2 . Workflow of DeBreak.(Chen Y. et al, 2023)
Figur2 . Workflow of DeBreak.(Chen Y. et al, 2023)
Install DeBreak
#bash conda create --name deb conda activate deb conda install -c bioconda debreak
General usage and parameters
debreak [-h] --bamSV caller for long-read sequencing data optional arguments: -h, --help Show this help message and exit -v, --version Show program's version number and exit --bam BAM Input sorted bam. index required --samlist SAMLIST A list of SAM files of same sample -o, --outpath OUTPATH Output directory --min_size MIN_SIZE Minimal size of detected SV --max_size MAX_SIZE Maxminal size of detected SV -d, --depth DEPTH Sequencing depth of this dataset -m, --min_support MIN_SUPPORT Minimal number of supporting reads for one event --min_quality MIN_QUALITY Minimal mapping quality of reads --aligner ALIGNER Aligner used to generate BAM/SAM -t, --thread THREAD Number of threads --rescue_dup Rescue DUP from INS calls. minimap2,ref required --rescue_large_ins Rescue large INS. wtdbg2,minimap2,ref required --poa POA for accurate breakpoint. wtdbg2,minimap2,ref required --no_genotype Disable genotyping -r, --ref REF Reference genome. Should be same with SAM/BAM --maxcov MAXCOV Maximal coverage for a SV. Suggested maxcov as 2 times mean depth --skip_detect Skip SV raw signal detection --tumor Allow clustered SV breakpoints during raw SV signal detection
Run SV Detection
DeBreak requires a input of read alignment results in BAM format. If you start with sequencing reads (Fasta or Fastq format), you may use minimap2 and samtools to map them to a reference genome before you can apply DeBreak:
#bash #minimap2 -a reference.fa movie1.fastq | samtools sort -o movie.aligned.sort.bam debreak --bam movie.aligned.sort.bam -o debreak_out/ --rescue_large_ins --rescue_dup --poa --ref hg38.fa
The output directory includes:
debreak.vcf Standard VCF file of SV calls. The chromosome, coordinates, size, type, number of supporting reads, mapping quality, genotype, and multi-allelic information are recorded for each SV call. debreak-allsv-merged-final Tab-delimited SV list, containing the name of reads supporting each SV call. sv_raw_calls/ Includes all SV raw signals on each chromosome. debreak_poa_workspace/ Temporary files during SV breakpoint refinement with POA. For debug purpose. debreak_ins_workspace/ Temporary files during ultra-large insertion detection. For debug purpose. map_depth/ Temporary files during sequencing depth estimation. For debug purpose.
Visualize in IGV
Import .vcf and .bam to visually inspect breakpoints and transcript context.
Part 3: Identifying Fusion Genes with FusionSeeker
FusionSeeker is optimized for long-read fusion detection, including complex or multi-breakpoint events.
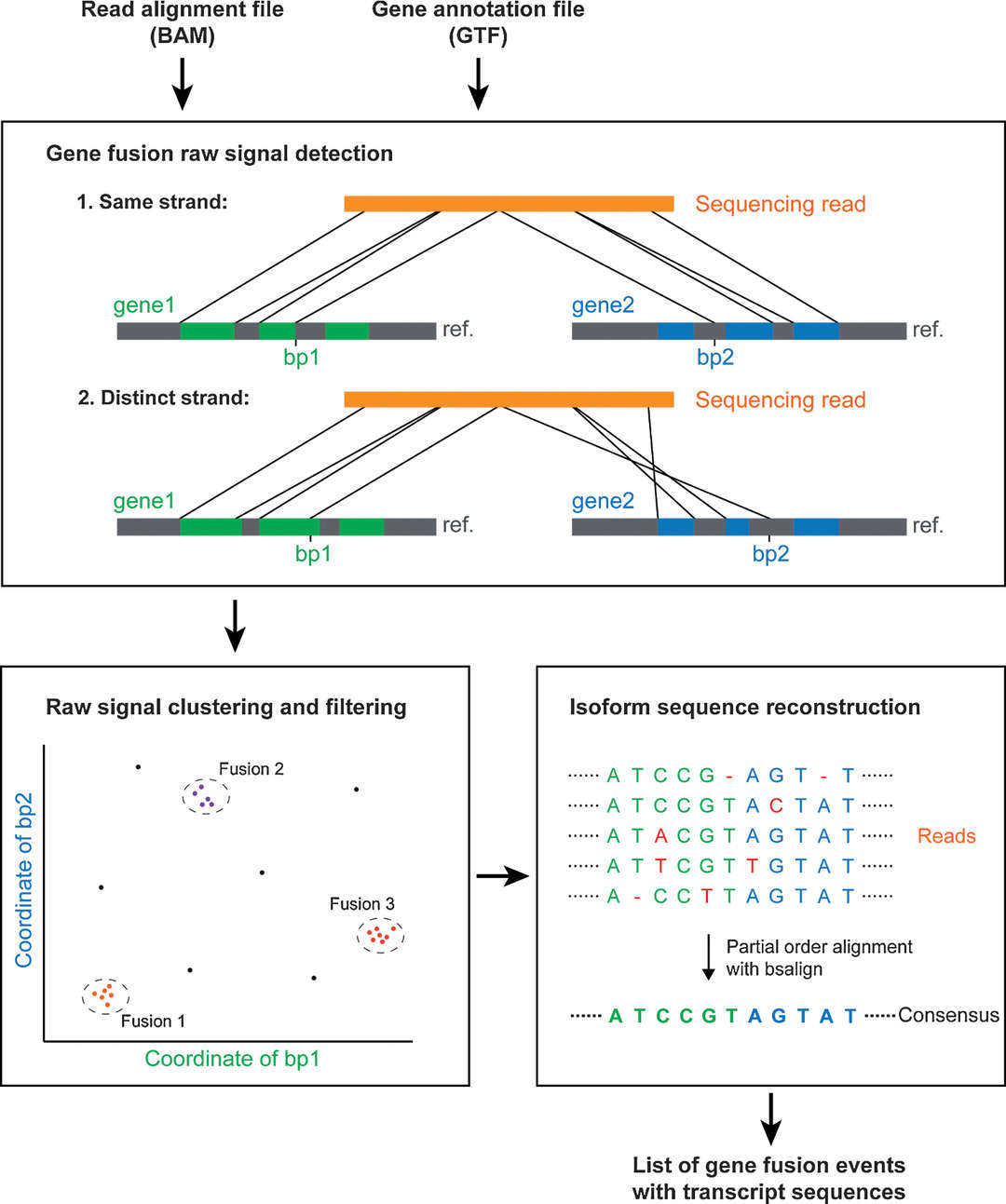 Figur3 . Workflow of FusionSeeker.(Chen Y. et al, 2023)
Figur3 . Workflow of FusionSeeker.(Chen Y. et al, 2023)
Install FusionSeeker
#bash git clone https://github.com/liulab-dfci/FusionSeeker.git cd FusionSeeker export PATH=$PWD/FusionSeeker/:$PATH conda create --name fusions -y conda activate fusions conda install -c bioconda minimap2=2.24 pysam=0.17 samtools=1.9 -y git clone https://github.com/ruanjue/bsalign.git cd bsalign && make export PATH=$PWD:$PATH
General usage and parameters
fusionseeker [-h] --bam Gene fusion caller for long-read sequencing data optional arguments: -h, --help show this help message and exit -v, --version show program's version number and exit --bam BAM Input sorted BAM. index required --datatype DATATYPE Input read type (isoseq, nanopore) [nanopore] --gtf GTF Genome annotation file --ref REF Reference genome. Required for breakpoint polishing --geneid Use Gene ID instead of Gene name [False] --human38 Use reference genome and GTF for Human GCRh38 (default) --human19 Use reference genome and GTF for Human GCRh37 -o OUTPATH, --outpath OUTPATH Output directory [./fusionseeker_out/] -s MINSUPP, --minsupp MINSUPP Minimal reads supporting an event [auto] --maxdistance MAXDISTANCE Maximal distance to cluster raw signals [20 for isoseq, 40 for nanopore] --keepfile Keep intermediate files [False] --thread THREAD Number of threads [8]
FusionSeeker Input
FusionSeeker requires a input of read alignment results in BAM format sorted by coordinates. If you start with sequencing reads (Fasta or Fastq format), you may use minimap2 and samtools to map them to a reference genome before you can apply FusionSeeker:
#bash #PacBio Iso-Seq minimap2 -ax splice:hq reference.fa isoseq.fastq | samtools sort -o isoseq.bam samtools index isoseq.bam
Gene Fusion Detection
fusionseeker --bam isoseq.bam --datatype isoseq -o fusionseeker_out/
The output directory includes:
confident_genefusion.txt A list of confident gene fusion calls from input BAM file. Includes gene names, breakpoint positions, number and name of fusion-supporting reads. confident_genefusion_transcript_sequence.fa Transcript sequences of reported confident gene fusion calls. clustered_candidate.txt A full list of gene fusion candidates before applying filters. rawsignal.txt A list of all gene fusion raw signals. log.txt Log file for debug. (raw_signal/ Intermediate files during raw signal detection. Removed by default.) (poa_workspace/ Intermediate files during transcript sequence generation with POA. Removed by default.)
Validate and Visualize
Use IGV to manually inspect junction reads and fusion boundaries.
Conclusion
Iso-Seq technology has ushered in a new era of transcriptomics by enabling direct, full-length sequencing of RNA molecules. Its ability to accurately identify alternative splicing, novel isoforms, structural variants, and gene fusions makes it a game-changer for complex transcriptome analysis. While it may involve higher cost and lower throughput than short-read sequencing, the quality and completeness of the data it delivers far outweigh these limitations, especially in research areas like cancer, neuroscience, and developmental biology.
As sequencing technologies continue to improve and become more accessible, the integration of Iso-Seq into standard transcriptome workflows is expected to become increasingly common. When combined with tools like IsoSeq3, DeBreak, and FusionSeeker, researchers can extract unparalleled insights into transcript structure and regulation-transforming our understanding of gene expression in health and disease.
Reference
- Ramberg S et al., A de novo Full-Length mRNA Transcriptome Generated From Hybrid-Corrected PacBio Long-Reads Improves the Transcript Annotation and Identifies Thousands of Novel Splice Variants in Atlantic Salmon. Front Genet. 2021 Apr 27;12:656334. https://doi.org/10.3389/fgene.2021.656334
- Chen Y et al., Deciphering the exact breakpoints of structural variations using long sequencing reads with DeBreak. Nat Commun. 2023 Jan 17;14(1):283. https://doi.org/10.1038/s41467-023-35996-1
- Chen Y et al., Human Genome Structural Variation Consortium; Chen H, Chong Z. Gene Fusion Detection and Characterization in Long-Read Cancer Transcriptome Sequencing Data with FusionSeeker. Cancer Res. 2023 Jan 4;83(1):28-33. https://doi.org/10.1158/0008-5472.CAN-22-1628
For research purposes only, not intended for personal diagnosis, clinical testing, or health assessment