Direct RNA Sequencing in Plants Reveals Isoforms, Poly(A) Tails, and Modifications
At a glance:
- Overcoming Short-Read Limitations in Plant Genomics with Direct RNA Sequencing (DRS)
- Direct RNA Sequencing: Capturing Full-Length Native RNA Molecules
- Applications of Direct RNA Sequencing (DRS) in Plant Research
- Innovative Discoveries with Direct RNA Sequencing (DRS) in Plants
Overcoming Short-Read Limitations in Plant Genomics with Direct RNA Sequencing (DRS)
Plant genomics and transcriptomics research has long faced the restrictions of short-read sequencing. Incomplete transcript assembly, poor detection of fusion genes, and the inability to capture RNA modifications have limited biological insight. These challenges often leave researchers with partial views of the plant transcriptome.
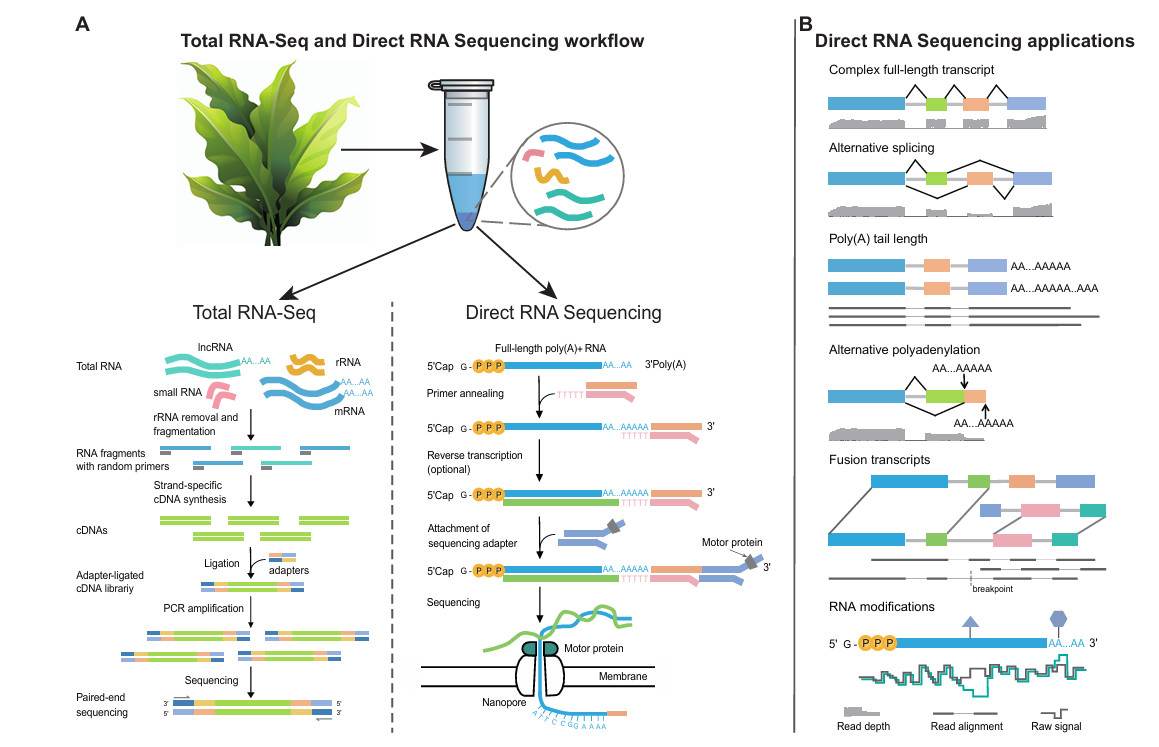
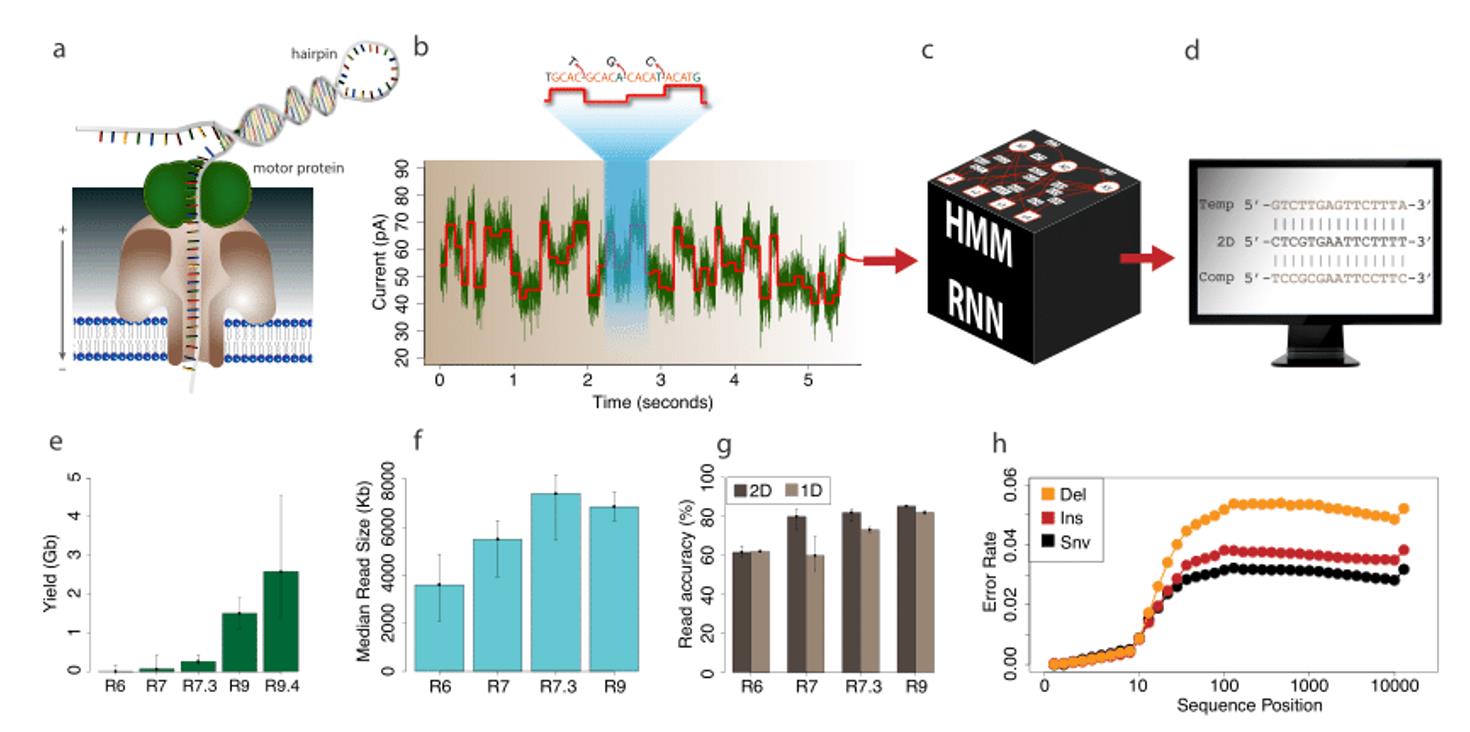
Direct RNA Sequencing (DRS), enabled by Oxford Nanopore technology, is rapidly transforming this field. Unlike traditional methods, DRS sequences RNA molecules directly without reverse transcription or amplification. This unique capability preserves full-length transcripts, native poly(A) tails, and chemical modifications. As a result, scientists gain an authentic view of transcript diversity and regulation.
Why DRS is a Game-Changer for Plant Studies
- Improved transcript resolution: Detects complete isoforms and alternative splicing patterns critical for plant stress responses.
- Fusion gene discovery: Identifies complex rearrangements that short-read technologies often miss.
- Epitranscriptomics unlocked: Captures modifications like m6A that regulate growth, development, and environmental adaptation.
- True-to-nature data: Avoids artefacts from cDNA conversion and amplification, ensuring higher biological accuracy.
By moving beyond the constraints of short-read sequencing, DRS opens a new era of plant transcriptomics. It enables crop scientists, molecular biologists, and agricultural R&D teams to explore RNA biology with unprecedented clarity.
Direct RNA Sequencing: Capturing Full-Length Native RNA Molecules
Direct RNA Sequencing (DRS), developed by Oxford Nanopore Technologies (ONT), is reshaping transcriptome research. Unlike conventional RNA-seq, which requires reverse transcription into cDNA and PCR amplification, DRS bypasses these steps entirely. Instead, it sequences complete, native RNA molecules in real time.
This innovation eliminates artefacts introduced during cDNA conversion and amplification, providing a more faithful view of transcript structure and regulation. By retaining full-length molecules, poly(A) tails, and chemical modifications, DRS enables researchers to study RNA biology as it truly exists within the cell.
Core Advantages of Direct RNA Sequencing (DRS)
1. No Reverse Transcription or PCR Amplification
DRS sequences native RNA molecules directly, eliminating the need for cDNA synthesis or PCR. This avoids the amplification bias and errors introduced by conventional workflows, ensuring data reflects the true biological state.
2. Full-Length Transcript Coverage
Instead of fragmented assembly, DRS captures the entire RNA sequence from the 5′ end to the poly(A) tail. This enables accurate identification of isoforms, alternative splicing events, and gene fusions without computational stitching.
3. Simultaneous Detection of RNA Modifications
The nanopore signal can sensitively detect chemical modifications such as m6A, m1A, m5C, m7G, hm5C, and Ψ. This "sequence-and-modify" capability allows multi-dimensional analysis, combining transcript identity with epitranscriptomic profiling.
4. High-Resolution Poly(A) Tail Analysis
DRS directly measures poly(A) tail length in parallel with isoform detection. This provides precise insights into alternative polyadenylation (APA) and its regulatory mechanisms in RNA stability, translation, and developmental control.
By combining these features, DRS offers a uniquely comprehensive view of RNA biology. It empowers researchers in plant science, biomedical studies, and drug discovery to capture transcript diversity, chemical regulation, and post-transcriptional control with unparalleled accuracy.
Service you may interested in
Applications of Direct RNA Sequencing (DRS) in Plant Research
Direct RNA Sequencing (DRS) provides plant scientists with insights beyond the reach of short-read sequencing. By reading full-length native RNA molecules directly, this technology supports a wide range of applications, from transcript discovery to epitranscriptomic mapping.
1. Full-Length Transcript Identification
DRS uncovers novel genes and isoforms without assembly errors, making it especially valuable for complex genomes such as polyploid and highly heterozygous plants.
2. Alternative Splicing Analysis
The method precisely captures splicing patterns and reveals tissue- or stress-specific splicing events that shape gene regulation.
3. Fusion Transcript Detection
DRS identifies transcripts formed between different genes or between genes and transposable elements, shedding light on genome structure variation and its functional impact.
4. Poly(A) Tail Length Measurement
By directly quantifying poly(A) tails, DRS allows researchers to investigate how tail length influences mRNA stability and translation efficiency.
5. Alternative Polyadenylation (APA) Mechanisms
The approach directly detects poly(A) site selection, enabling analysis of APA events and their roles in fine-tuning gene expression.
6. RNA Modification Mapping
At a transcriptome-wide scale, DRS pinpoints RNA modifications such as m6A and m5C. These epitranscriptomic maps reveal how chemical marks regulate plant development, stress adaptation, and gene expression.
Innovative Discoveries with Direct RNA Sequencing (DRS) in Plants
Direct RNA Sequencing (DRS) has become a powerful tool for uncovering new transcriptome complexity in plants. By sequencing native RNA molecules directly, DRS enables researchers to capture isoforms, RNA modifications, and regulatory events with unprecedented clarity. Recent plant studies demonstrate its impact across species ranging from fast-growing bamboo to staple crops like rice.
Moso Bamboo (Phyllostachys edulis)
Expanded transcriptome annotation: Using DRS, researchers identified 22,360 novel transcripts, providing a more complete framework for studying rapid bamboo growth (Li et al., 2023).
Dynamic alternative polyadenylation (APA): Distinct APA site usage was observed between basal and middle tissues of bamboo shoots, suggesting a role in growth regulation (Li et al., 2023).
m6A modification maps: Stage-specific m6A marks were revealed, showing RNA methylation as a regulatory layer in development (Li et al., 2023).
CircRNA methylation: About 11% of circRNAs showed m6A near their backsplice junctions, linking RNA modifications to circRNA formation and stability (Wang et al., 2020).
High-confidence circRNA discovery: Improved library preparation combined with DRS identified 470 circRNAs, many linked to chromosomal organisation and segregation (Wang et al., 2020).
Arabidopsis thaliana
Hidden complexity uncovered: Even in this well-annotated model plant, DRS revealed 38,500 novel isoforms and 8,700 unique splicing variants (Parker et al., 2020; Zhang et al., 2020).
Precise splicing analysis: For the flowering regulator FLM, DRS identified 19 isoforms, including 11 previously unknown variants (Parker et al., 2020).
Poly(A) tail diversity: Transcriptome-wide analysis showed organ-specific tail length patterns, with notably longer tails in pollen and seeds, hinting at tissue-specific RNA stability regulation (Parker et al., 2020).
Functional validation of m6A: Loss of methylation led to defects in 3′ end processing and reduced transcript abundance, confirming m6A's essential role in RNA maturation (Parker et al., 2020).
Novel methyltransferase discovery: FIOT1, a METTL16 homologue, was identified as a new m6A writer in Arabidopsis. Mutants showed global m6A reduction and early flowering (Xu et al., 2022).
TE-gene chimeras: DRS pinpointed ~3,000 transcripts formed from transposon-gene fusions, showing how TE insertions modulate host gene expression under stress (Bertheller et al., 2023).
Rice (Oryza sativa) and Wild Relatives (Oryza spp.)
Fusion transcript evolution: Comparative studies across Oryza species identified 310 fusion transcripts. Although frequently formed, few became fixed, showing strong purifying selection—supporting the idea that fusions can spark new gene origins (Zhou et al., 2022).
Tissue-specific m6A maps: Across six tissues, 276–2,042 transcripts carried distinct methylation patterns, linking m6A to rice tissue differentiation and specialised functions (Yu et al., 2023a).
m5C modification under stress: In salt-stressed plants, downregulated genes enriched for m5C were linked to stress-response pathways, suggesting RNA methylation as a key regulator of resilience (Wu et al., 2024).
Polyploid Crops
Brassica napus (rapeseed): DRS revealed intricate alternative splicing patterns in this allopolyploid species compared with its diploid parents (Brassica rapa and Brassica oleracea). These findings highlight transcriptome reprogramming as a key factor in polyploid adaptation (Li et al., 2022).
London plane (Platanus × acerifolia): In dodecaploid trees, researchers detected 1,854 unique splicing events absent in hexaploid relatives. These transcripts were enriched in DNA repair pathways, suggesting a role in maintaining genome stability at higher ploidy levels (Yan et al., 2023).
Forestry Species
Populus trichocarpa (black cottonwood): Under drought stress, xylem cambial cells shifted toward distal poly(A) site usage, generating longer 3′ UTRs and extended poly(A) tails. This post-transcriptional strategy may fine-tune stress adaptation. Integrating DRS with proteomics further revealed coordinated changes from RNA methylation (m6A) to protein abundance (Gao et al., 2022).
Pine: Nutrient stress experiments showed that changes in alternative polyadenylation coincided with increased m6A deposition. This coupling highlights a synergistic role of RNA modification and 3′ end processing in nutrient response (Ortigosa et al., 2022).
Horticultural Crops
Strawberry: DRS uncovered 110,888 novel transcripts during fruit development. The white-fruit stage emerged as a critical transition point, where many unreported transcripts were expressed, shaping fruit quality traits (Chen et al., 2022).
Citrus species: Across nine citrus genomes, researchers predicted thousands of new transcripts and identified 2,613–3,389 novel long non-coding RNAs (lncRNAs). Comparative analysis revealed isoform variation among homologous genes, offering new insights into citrus domestication and evolution (Hu et al., 2022).
Cynara cardunculus (cardoon): DRS corrected 13,039 gene models derived from short-read assemblies. This improved gene and isoform annotations by 15% and 18% respectively, significantly enhancing genome quality for this non-model species (Puglia et al., 2020).
Cereal Crops
Wheat: Early seed development was shown to be rich in lncRNAs and transposon-derived transcripts. Researchers identified 796 novel lncRNAs, many containing retrotransposon sequences, underscoring the role of transposable elements in shaping complex transcriptomes (Kirov et al., 2020).
Maize: By combining DRS with polysome profiling, scientists found m6A enrichment in 3′ UTRs and strong correlation with distal poly(A) site usage. Transcripts carrying m6A marks displayed reduced translation efficiency, positioning m6A as a translational brake in maize (Luo et al., 2020).
Medicinal Plants
Camptotheca acuminata: DRS refined transcriptome annotations by correcting 5,692 splicing events across 4,746 genes. Importantly, 15 transcription factors were linked to regulation of camptothecin biosynthesis, offering new targets to boost production of this anti-cancer compound through synthetic biology (Zhang et al., 2023).
By spanning polyploid crops, forest trees, fruit species, cereals, and medicinal plants, DRS provides a unified framework for exploring RNA complexity. Its ability to capture isoforms, RNA modifications, and alternative polyadenylation equips researchers with tools to link transcript regulation to agronomic traits, stress adaptation, and secondary metabolite production.
References
- Zhu XT, Sanz-Jimenez P, Ning XT, Tahir Ul Qamar M, Chen LL. Direct RNA sequencing in plants: Practical applications and future perspectives. Plant Commun. 2024;5(11):101064. doi:10.1016/j.xplc.2024.101064
- Berthelier, J., Furci, L., Asai, S., Sadykova, M., Shimazaki, T., Shirasu, K., and Saze, H. (2023). Long-read direct RNA sequencing reveals epigenetic regulation of chimeric gene-transposon transcripts in Arabidopsis thaliana. Nat. Commun. 14:3248.
- Chen Q., Lin X., Tang W., Deng Q., Wang Y., Lin Y., He W., Zhang Y., Li M., Luo Y., et al. Transcriptomic complexity in strawberry fruit development and maturation revealed by nanopore sequencing. Front. Plant Sci. 2022;13:872054. doi: 10.3389/fpls.2022.872054.
- Gao Y., Liu X., Jin Y., Wu J., Li S., Li Y., Chen B., Zhang Y., Wei L., Li W., et al. Drought induces epitranscriptome and proteome changes in stem-differentiating xylem of Populus trichocarpa. Plant Physiol. 2022;190:459–479. doi: 10.1093/plphys/kiac272.
- Hu X.L., You C., Zhu K., Li X., Gong J., Ma H., Sun X. Nanopore long-read RNAseq reveals transcriptional variations in citrus species. Front. Plant Sci. 2022;13:1077797. doi: 10.3389/fpls.2022.1077797.
- Kirov I., Dudnikov M., Merkulov P., Shingaliev A., Omarov M., Kolganova E., Sigaeva A., Karlov G., Soloviev A. Nanopore RNA sequencing revealed long non-coding and ltr retrotransposon-related RNAs expressed at early stages of triticale seed development. Plants. 2020;9:1794. doi: 10.3390/plants9121794.
- Li M., Hu M., Xiao Y., Wu X., Wang J. The activation of gene expression and alternative splicing in the formation and evolution of allopolyploid Brassica napus. Hortic. Res. 2022;9:uhab075. doi: 10.1093/hr/uhab075.
- Li T., Wang H., Zhang Y., Wang H., Zhang Z., Liu X., Zhang Z., Liu K., Yang D., Zhang H., Gu L. Comprehensive profiling of epigenetic modifications in fast-growing Moso bamboo shoots. Plant Physiol. 2023;191:1017–1035. doi: 10.1093/plphys/kiac525.
- Luo J.H., Wang Y., Wang M., Zhang L.Y., Peng H.R., Zhou Y.Y., Jia G.F., He Y. Natural variation in RNA m(6)a methylation and its relationship with translational status. Plant Physiol. 2020;182:332–344. doi: 10.1104/pp.19.00987.
- Ortigosa F., Lobato-Fernández C., Pérez-Claros J.A., Cantón F.R., Ávila C., Cánovas F.M., Cañas R.A. Epitranscriptome changes triggered by ammonium nutrition regulate the proteome response of maritime pine roots. Front. Plant Sci. 2022;13:1102044. doi: 10.3389/fpls.2022.1102044.
- Parker M.T., Knop K., Sherwood A.V., Schurch N.J., Mackinnon K., Gould P.D., Hall A.J., Barton G.J., Simpson G.G. Nanopore direct RNA sequencing maps the complexity of Arabidopsis mRNA processing and m(6)A modification. Elife. 2020;9:e49658. doi: 10.7554/eLife.49658.
- Puglia G.D., Prjibelski A.D., Vitale D., Bushmanova E., Schmid K.J., Raccuia S.A. Hybrid transcriptome sequencing approach improved assembly and gene annotation in Cynara cardunculus (L.) BMC Genom. 2020;21:317. doi: 10.1186/s12864-020-6670-5.
- Wang Y., Zou Q., Li F., Zhao W., Xu H., Zhang W., Deng H., Yang X. Identification of the cross-strand chimeric RNAs generated by fusions of bi-directional transcripts. Nat. Commun. 2021;12:4645. doi: 10.1038/s41467-021-24910-2.
- Wu Y., Shao W., Yan M., Wang Y., Xu P., Huang G., Li X., Gregory B.D., Yang J., Wang H., Yu X. Transfer learning enables identification of multiple types of RNA modifications using nanopore direct RNA sequencing. Nat. Commun. 2024;15:4049. doi: 10.1038/s41467-024-48437-4.
- Xu T., Wu X., Wong C.E., Fan S., Zhang Y., Zhang S., Liang Z., Yu H., Shen L. Fiona1-mediated m(6)A modification regulates the floral transition in Arabidopsis. Adv. Sci. 2022;9:e2103628. doi: 10.1002/advs.202103628.
- Yan X., Chen X., Li Y., Li Y., Wang F., Zhang J., Ning G., Bao M. The abundant and unique transcripts and alternative splicing of the artificially autododecaploid london plane (platanus x acerifolia) Int. J. Mol. Sci. 2023;24:14486. doi: 10.3390/ijms241914486.
- Yu F., Qi H., Gao L., Luo S., Njeri Damaris R., Ke Y., Wu W., Yang P. Identifying RNA Modifications by Direct RNA Sequencing Reveals Complexity of Epitranscriptomic Dynamics in Rice. Dev. Reprod. Biol. 2023;21:788–804. doi: 10.1016/j.gpb.2023.02.002.
- Zhang H., Li G., Fu C., Duan S., Hu D., Guo X. Genome-wide identification, transcriptome analysis and alternative splicing events of Hsf family genes in maize. Sci. Rep. 2020;10:8073. doi: 10.1038/s41598-020-65068-z.
- Zhang H., Shen X., Sun S., Li Y., Wang S., Wei J., Guo B., Sun C. Integrated transcriptome and proteome analysis provides new insights into camptothecin biosynthesis and regulation in Camptotheca acuminata. Physiol. Plant. 2023;175:e13916. doi: 10.1111/ppl.13916.
- Zhou Y., Zhang C., Zhang L., Ye Q., Liu N., Wang M., Long G., Fan W., Long M., Wing R.A. Gene fusion as an important mechanism to generate new genes in the genus Oryza. Genome Biol. 2022;23:130. doi: 10.1186/s13059-022-02696-w.

For research purposes only, not intended for personal diagnosis, clinical testing, or health assessment