Co-evolution of Cancer Genome and Epigenome: Insights into Tumor Adaptation and Treatment Resistance
Cancer is complicated not only because of all the mutations that build up over time but also because the genome and epigenome are always changing together. This process, through a series of changes, gives cancer cells the ability to adapt and survive. It allows them to change their characteristics, avoid being detected by the immune system, and resist drugs used to treat them.
In traditional oncology, hard mutations in the genome are often seen as targets for treatment. However recent studies have shown that soft changes in epigenetic regulation (like reprogramming of DNA methylation and dynamic chromatin remodeling) also drive tumor evolution and can even lead to the accumulation of genetic mutations. The buildup of genetic mutations creates a kind of "pre-adaptive environment." For example, chemotherapy can cause problems with the body's ability to repair DNA, and it can also make cancer cells change more quickly. In some cases, a lack of oxygen can help the body find cancer cells that are more likely to grow and spread. Systems biology is a field that uses math and computer science to study complex networks. It has developed new tools to analyze this network. These tools are helping scientists understand how different cells and genes work together. They are also helping scientists find new ways to treat cancer.
This paper provides an in-depth analysis of how cancer cells adapt to therapeutic pressure, immune escape and metastatic spread through the synergistic evolutionary mechanism of gene mutation and epigenetic regulation from a systems biology perspective, revealing new targets and strategic directions for precision therapy.
Genome-Epigenome Co-Evolution
Cancer evolution is the result of dynamic interactions between the genome and epigenome. Genomic mutations provide genetic diversity, while epigenetic regulation provides phenotypic plasticity to cancer cells through changes in chromatin and DNA methylation. The synergy between the two allows for sustained tumor adaptation in response to microenvironmental stresses and therapeutic interventions. This section will explore how mutational stress and cancer stem cell-like states work together to drive this process.
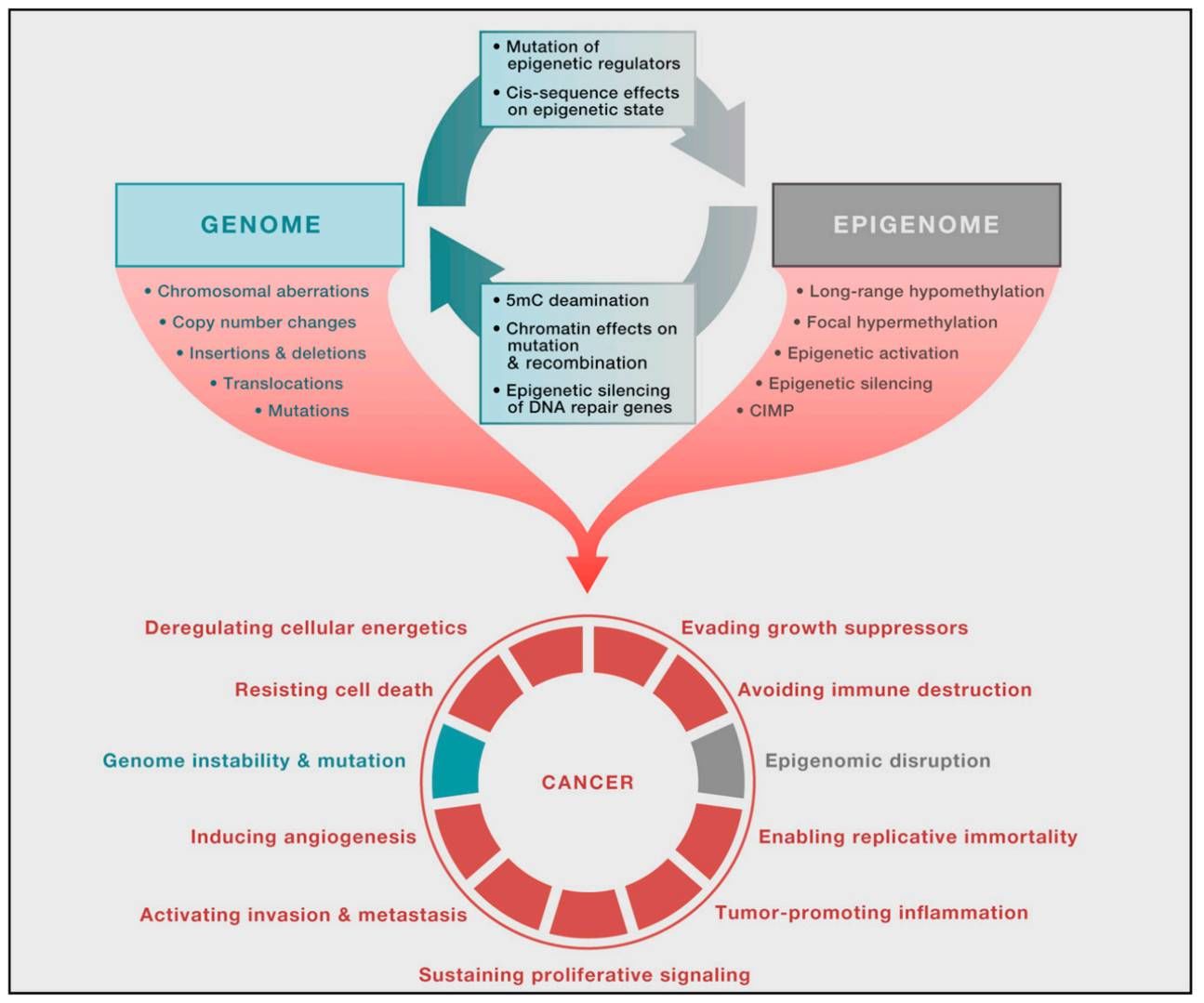 Interplay between the cancer genome and epigenome (Shen et al., 2013)
Interplay between the cancer genome and epigenome (Shen et al., 2013)
Service you may intersted in
Learn More
Mutational stress and epigenetic plasticity
Mutational stress is caused by the genetic instability of cancer cells when they are under duress. Stresses such as chemotherapy or hypoxia can activate error-prone repair mechanisms (e.g., trans-damage synthetic polymerases), leading to the formation of localized mutation clusters. For instance, the hypoxic microenvironment characteristic of glioblastoma has been shown to reduce methylation levels of DNA repair genes, such as MLH1, leading to a significant increase in mutation rates. Metabolic reprogramming further exacerbates this process. Lactate produced by the Watt's effect inhibits histone deacetylases (HDACs), leading to increased chromatin loosening and replication errors. Elevated levels of α-ketoglutarate (α-KG) activate the TET enzyme in KRAS mutation-driven pancreatic cancers, creating a positive feedback loop of mutation and epigenetic modification.
Clinical intervention strategies are evolving to focus on addressing the source of mutational stress. Studies on melanoma have demonstrated that the intermittent administration of BRAF inhibitors can reduce the likelihood of resistance to stress-induced mutations. Furthermore, the use of epimeric agents, such as DNA methyltransferase (DNMT) inhibitors, has been shown to enhance chemosensitivity by restoring the silenced state of DNA repair genes. These advances underscore the significance of targeting both the genome and the epigenome simultaneously.
Cancer Stem Cell States and Evolutionary Fitness
The epigenetic plasticity of cancer stem cells (CSCs) is critical for maintaining their evolutionary advantage. For instance, CD44+ breast cancer stem cells display a "biphasic pattern" of global DNA hypomethylation and hypermethylation of differentiated genes, thereby facilitating a shift in phenotypes through the activation of the Wnt pathway and the suppression of Notch signaling. Single-cell analysis revealed that the chromatin open regions of CSCs were enriched with pluripotency factor binding sites, forming a stable episodic attractor state.
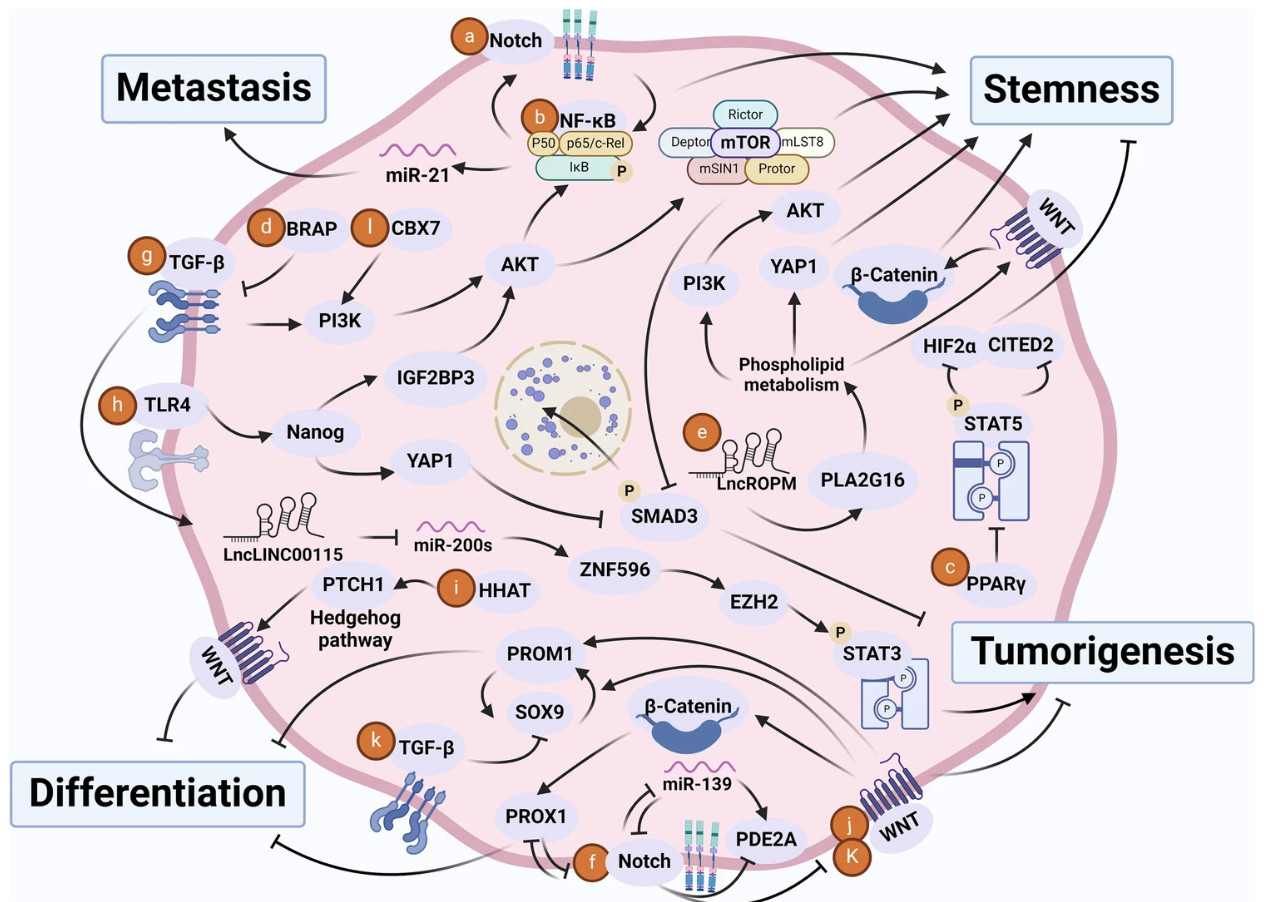 Crosstalk of signaling pathways in CSCs (Chu et al., 2024)
Crosstalk of signaling pathways in CSCs (Chu et al., 2024)
The tumor microenvironment (e.g., hypoxia) has been shown to influence the adaptation of CSCs through episodic memory mechanisms.HIF-1α has been observed to induce glycolysis, as well as to inhibit DNMT3A activity and lower the methylation threshold of oncogenes. Cyclic hypoxia in glioblastoma triggers a dynamic modification of H3K27ac, contributing to the transformation of CSCs to an aggressive phenotype. Conventional chemotherapy has difficulty removing CSCs because it relies on epigenetically regulated resistance mechanisms (e.g., ABC transporter pumps). Recent studies have shown that combining EZH2 inhibitors with immunotherapy has led to a significant reduction in the proportion of CSCs in lung cancer models. This suggests new directions for targeting evolutionary adaptations in lung cancer treatment.
Adaptive Strategies in Co-Evolution
Cancer evolution is synergistically driven by the genome and epigenome. Under therapeutic pressure, cancer cells undergo chromatin dynamic reprogramming to achieve short-term phenotypic adaptation while screening for genetic mutations to consolidate long-term survival advantages. This "dual-engine" mechanism enables tumors to exhibit robust adaptations in immune escape and metastasis. In this chapter, we analyze the systematic evolutionary strategy of cancer from two dimensions: chromatin response and immune regulation.
Chromatin Dynamics under Therapeutic Stress
Chromatin homeostasis is directly perturbed by chemotherapy or targeted therapies.DNA damage activates the ATM/ATR pathway, leading to chromatin relaxation and initiating the expression of repair genes, but solid tumors can utilize integrin-YAP signaling to stretch chromatin and activate pro-invasive genes (e.g., LOX). Episodic memory plays a key role in this process: short-term stress induces H3K4me3 enrichment at stress gene promoters, whereas long-term treatment triggers global loss of H3K27me3 and promotes stem cell properties. Single-cell polytomies revealed that residual cancer cells "target" drug-resistant genes (e.g., ABCB1) through DNA methylation, resulting in persistent treatment resistance. Current technologies such as CRISPR-dCas9 epitope editing can reverse drug resistance modifications, while spatial multi-omics resolves microenvironmental and chromatin interactions.
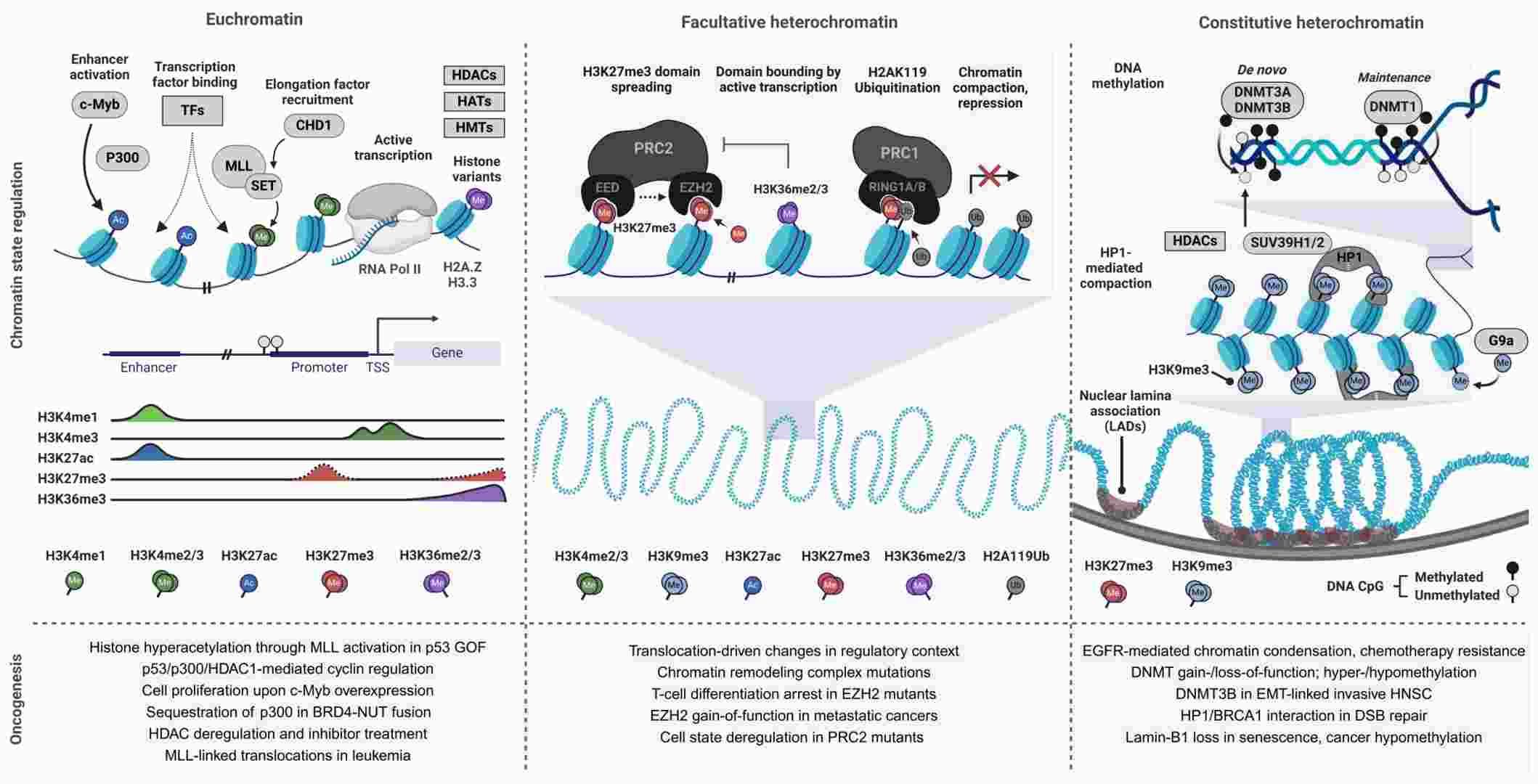 Chromatin state regulation and misregulation in cancer (Kiri & Tyrone, 2024)
Chromatin state regulation and misregulation in cancer (Kiri & Tyrone, 2024)
Immune Evasion and Metastatic Potential
Cancer evades immune recognition by episodically silencing MHC-I antigen presentation (e.g., HLA-A/B hypermethylation). Demethylating drugs combined with PD-1 inhibitors restore antigen presentation and enhance immune efficacy. Lactate accumulation in the microenvironment activates PD-L1 through histone acetylation (H3K18la), forming a "metabolic-immune" escape axis, which is validated by inhibitors targeting LDH. During metastasis, EMT episodic switches (e.g., SNAI1 recruits DNMT3A to silence E-cadherin) and non-coding RNA networks (e.g., miR-200/ZEB1 loop) drive cellular plasticity. Circulating tumor cells activate dormant genes (NR2F1) via H3K4me1 modification to form pre-metastatic ecological niches in distal organs. In the clinic, ctDNA methylation markers (e.g. SEPTIN9) dynamically monitor epigenetic clonal evolution.
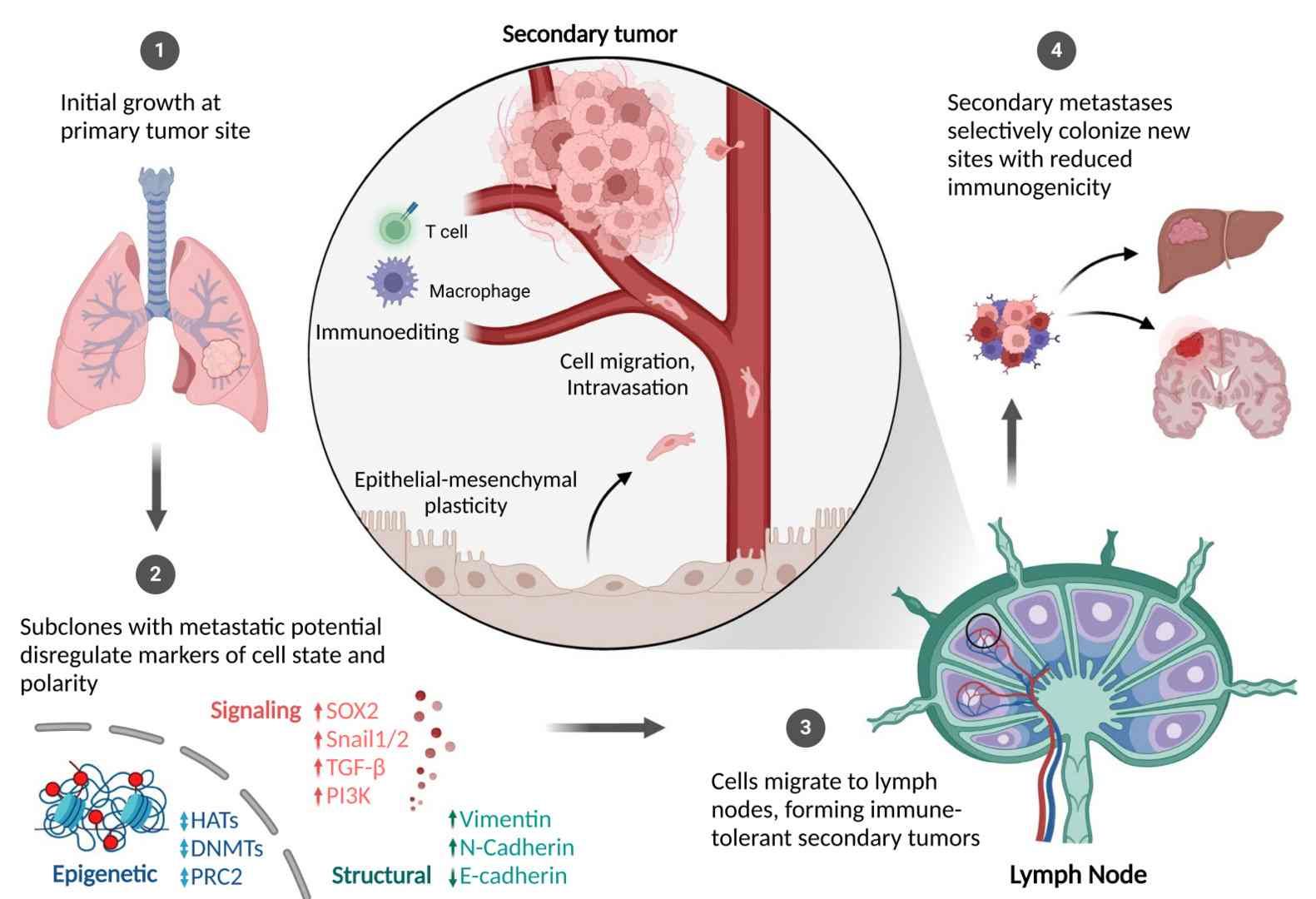 Cell state deregulation, migration, and metastasis (Kiri & Tyrone, 2024)
Cell state deregulation, migration, and metastasis (Kiri & Tyrone, 2024)
Co-Evolutionary Signatures in Cancer Progression
The synergistic evolution of the cancer genome and epigenome is a central driver of malignant tumor progression. They work together to shape the adaptive phenotype of cancer cells through dynamic interactions: genetic variation triggers global epigenetic remodeling, while epigenetic regulation provides targets for subsequent mutation accumulation. From a systems biology perspective, this synergy forms an "environment-metabolism-epigenetic-genetic" cascade feedback network that enables cancer cells to respond flexibly to therapeutic pressures. In this section, the molecular logic and clinical significance of the co-evolutionary features of cancer will be revealed by analyzing the evolutionary state and the integrated regulatory network of cellular plasticity from the perspective of multi-omics technology.
Multi-Omics Profiling of Evolutionary States
With breakthroughs in single-cell technology and spatial histology, researchers have been able to capture cancer evolutionary trajectories at single-cell resolution. For example, single-cell multi-omics technologies (e.g., BD Rhapsody) allow simultaneous analysis of the transcriptome, epigenome, and proteome of the same cell, revealing the heterogeneity of tumor subclones. In colorectal cancer, mutations in the APC gene often coincide with enhanced chromatin accessibility in the regulatory regions of the Wnt pathway, suggesting that epigenetic remodeling may provide a "pre-adaptive" environment for genetic variation. Spatial multi-omics (e.g., 10x Visium) further revealed spatial co-localization of H3K27ac activation markers and EGFR amplification events in hypoxic regions of tumors, suggesting that microenvironmental stresses may drive both epigenetic and genetic alterations.
Deep learning models provide new tools for multi-omics data integration. Algorithms such as Subtype-DCC integrate transcriptomic and methylation data through comparative learning to achieve subtype-accurate classification in breast cancer (AUC of 0.92) and identify epigenetic-genetic covariant modules associated with chemoresistance. Dynamic trajectory analysis techniques (e.g., RNA Velocity), on the other hand, reconfigured cancer cell state transition pathways, such as during EMT, where temporal up-regulation of ZEB1 and episodic silencing of the miR-200 family formed a negative feedback loop to drive cellular plasticity transition.
Integrative Landscapes of Cancer Cell Plasticity
Cellular plasticity is a phenotypic output of genomic and epigenomic co-evolution, with regulatory networks spanning microenvironmental signaling, metabolic reprogramming, and epigenetic-genetic interactions. For example, TGF-β1 secreted by tumor-associated fibroblasts (CAFs) induces EMT while enhancing cancer stem cell properties through the miR-200/ZEB1 loop. In this process, transcription factors such as SNAI1 recruit DNMT3A and HDAC1 to silence epithelial genes (e.g., E-cadherin), while altered chromatin accessibility further promotes the expression of pro-metastatic genes (e.g., LOX, MMP).
Metabolic-epigenetic coupling is another key mechanism. Fluctuations in acetyl coenzyme A levels regulate histone acetylation and affect Wnt/β-catenin pathway activity; whereas, when SAM metabolism is restricted, reduced H3K27me3 modification leads to de-repression of pluripotency genes (e.g., OCT4) and promotes dedifferentiation. Computational models (e.g., MOSA) integrating multi-omics data found that glutamine metabolism was significantly associated with high DNMT1 expression, suggesting that metabolic interventions may reverse epigenetically mediated chemoresistance.
Therapeutic Targeting of Co-Evolution
Co-evolution of the cancer genome and epigenome constitutes a central mechanism of tumor heterogeneity and treatment resistance. This dynamic process enables cancer cells to continuously adapt to therapeutic stresses through a "mutation-epigenome-microenvironment" cascade. For example, epigenetic remodeling (e.g., DNA hypomethylation or enhancer reprogramming) not only drives mutation accumulation but also mediates the screening of drug-resistant clones. This section explores how to target co-evolutionary networks to achieve therapeutic breakthroughs in terms of both epigenetic vulnerability interventions and precision medicine applications.
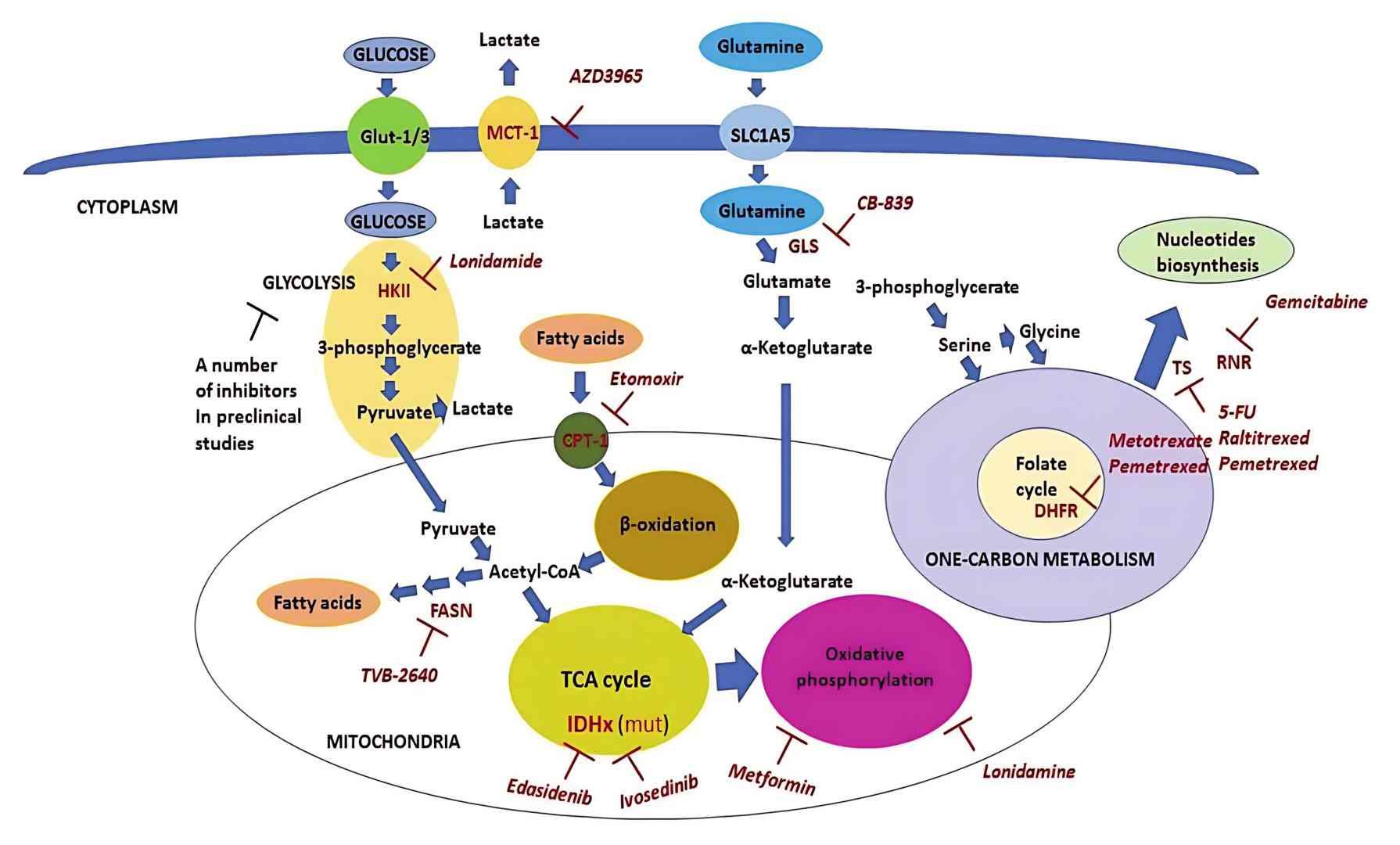 The scheme of metabolic pathways which are targeted in clinical oncology (Shuvalov et al., 2021)
The scheme of metabolic pathways which are targeted in clinical oncology (Shuvalov et al., 2021)
Intervention Strategies for Epigenomic Vulnerabilities
The plasticity of epigenetic regulation provides unique targets for cancer therapy. Epigenetic alterations, such as aberrant DNA methylation and imbalanced histone modifications, are both drivers of tumorigenesis and critical nodes for maintaining cancer cell survival. Significant progress has been made in the development of drugs against these targets: DNMT inhibitors restore the expression of oncogenes by reversing their hypermethylation; EZH2 inhibitors (e.g., Tazemetostat) block H3K27me3-mediated gene silencing; and BRD4 inhibitors (e.g., JQ1) exert their therapeutic efficacy by interfering with oncogene transcription.
Combination therapy strategies can overcome the limitations of monotherapy. The combination of epigenetic drugs with chemotherapy and immunotherapy has shown synergistic effects: DNMT inhibitors in combination with PD-1 blockers activate endogenous retroviral signaling and enhance the immune response, while EZH2 inhibitors in combination with PARP inhibitors amplify the DNA damage response through epigenetic modulation. Such strategies significantly delay the emergence of drug resistance through synergistic blockade of multiple pathways.
Precision Oncology via Co-Evolutionary Insights
Precision oncology is evolving from "static typing" to "dynamic tracking". The integration of multi-omics technologies and the application of artificial intelligence models enable clinics to analyze the trajectory of tumor evolution in real-time and predict the response to treatment. For example, spatial transcriptome analysis can identify enhancer reprogramming events in colorectal cancer that predate gene mutations, guiding early intervention; single-cell epigenome monitoring can predict the risk of osteosarcoma recurrence in advance by tracking dynamic changes in H3K9ac.
Artificial intelligence and gene editing technologies further promote individualized treatment. Deep learning models integrate genomic mutations, epigenetic modifications, and clinical data to improve the accuracy of epigenetic drug response prediction to 82%; CRISPR epigenetic editing technology can target the repair of promoter methylation of oncogenes (e.g., MLH1) to reverse immunotherapy resistance in colorectal cancer.
References
- Shen, Hui, and Peter W Laird. "Interplay between the cancer genome and epigenome." Cellvol. 153,1 (2013): 38-55. doi:10.1016/j.cell.2013.03.008
- Chu, Xianjing et al. "Cancer stem cells: advances in knowledge and implications for cancer therapy." Signal transduction and targeted therapy vol. 9,1 170. 5 Jul. 2024, doi:10.1038/s41392-024-01851-y
- Kiri, Saurav, and Tyrone Ryba. "Cancer, metastasis, and the epigenome." Molecular cancer vol. 23,1 154. 2 Aug. 2024, doi:10.1186/s12943-024-02069-w
- Shuvalov, Oleg et al. "Linking Metabolic Reprogramming, Plasticity and Tumor Progression." Cancers vol. 13,4 762. 12 Feb. 2021, doi:10.3390/cancers13040762




