Chromatin Interaction Capture Technology Comparison: From Hi-C to Next-Generation HiChIP and PLAC-seq for 3D Genome Analysis
At a glance:
- The Three-Dimensional Challenge: Why 1D Genomics Fails to Resolve Chromatin Interaction
- Chromatin Interaction Technology Overview: Global vs. Protein-Centric Focus
- Service you may intersted in
- From ChIA-PET to HiChIP: The Evolution of Protein-Centric Capture
- Detailed Comparative Analysis: Optimizing Your Chromatin Interaction Strategy
- Functional Applications in Research and Disease Modeling
- Advanced Bioinformatics for 3D Genomic Data Integrity
- Conclusion and Recommendations
- Selecting Your 3D Genome Strategy and Next Steps
- Frequently Asked Questions (FAQs)
The Three-Dimensional Challenge: Why 1D Genomics Fails to Resolve Chromatin Interaction
The linear sequence of DNA is only half the story in advanced gene regulation. Genome conformation is central to gene control, dictating when and how genes are expressed.
1D sequencing assays, such as ChIP-seq, effectively map the binding sites of transcription factors or histone marks. However, these methods only provide one-dimensional localization data. They inherently fail to resolve the functional, long-range regulatory connections that define cellular state.
The human genome's two-meter-long DNA molecule is intricately folded within the nucleus. Capturing the physical contact between physically distant regulatory elements is the key technical challenge. This communication occurs via Chromatin Loops, often linking enhancers to their target promoters.
To fully map the functional 3D Genome architecture, researchers must move beyond linear analysis. Specialized methodologies are required to isolate, capture, and sequence these specific three-dimensional contacts. This approach provides authoritative data for dissecting transcriptional control mechanisms and disease etiology.
This transition in methodology is achieved through advanced Chromosome Conformation Capture (3C) techniques.

Chromatin Interaction Technology Overview: Global vs. Protein-Centric Focus
The field of Chromosome Conformation Capture (3C) has evolved significantly to meet the demands of 3D Genome research. These techniques are broadly categorized by their experimental scope: global mapping or protein-centric capture. Choosing the correct strategy depends on whether the goal is to map the entire folding structure or just the interactions mediated by a specific factor.
The Global Perspective: Hi-C and 4C-seq
Global methods provide a comprehensive look at the entire 3D Genome architecture.
- Hi-C Sequencing (Global): Hi-C is the foundational tool for surveying all Chromatin Interaction within the genome in a single experiment. This approach provides an unbiased, genome-wide view of how chromatin is organized, enabling the detailed characterization of large-scale structures like Topologically Associating Domains (TADs) and Chromatin Loops. Hi-C data is essential for understanding the spatial organization that influences genome stability and function.
- Recommended Reading: Learn more about our high-resolution.
- 4C-seq (Locus-Specific): Also known as circular 3C, 4C-seq allows researchers to identify all genomic regions that interact with a single, predetermined point of interest, known as the "viewpoint". This method is highly effective for focusing analysis on a specific promoter or regulatory element and finding all its interacting partners.
The Protein-Centric Approach: Enhanced Specificity
While Hi-C reveals the structure, it does not directly identify the agents—the proteins—responsible for that structure. To achieve functional specificity, techniques incorporate an essential Immunoprecipitation (IP) step.
This IP step, also known as ChIP, enriches for DNA fragments bound by the specific protein of interest (e.g., CTCF, Cohesin, or H3K27ac). By adding this critical focus, protein-centric methodologies drastically reduce sample complexity. This focuses sequencing capacity precisely on the functionally relevant Chromatin Loops mediated by the targeted factor.
The two main generations of protein-centric methods are ChIA-PET and the highly efficient next-generation methods, HiChIP and PLAC-seq. This focus results in a superior signal-to-noise ratio, moving analysis from simple structural mapping to mechanistic discovery.
Service you may intersted in
From ChIA-PET to HiChIP: The Evolution of Protein-Centric Capture
The drive for higher efficiency and lower sample input has rapidly advanced protein-centric mapping. This evolution culminated in the streamlined, high-sensitivity methods we offer today.
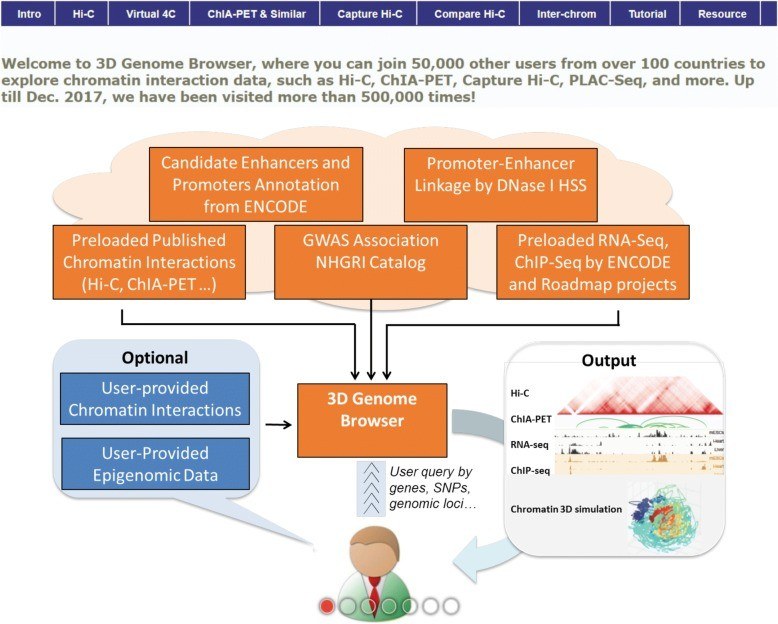 Fig 2. The overall design of the 3D Genome Browser
Fig 2. The overall design of the 3D Genome Browser
ChIA-PET: The Pioneer Technology
Chromatin Interaction Analysis by Paired-End Tag sequencing (ChIA-PET) was the first method to successfully combine Chromatin Immunoprecipitation (ChIP) with Chromatin Interaction Linking. This technique enabled the early mapping of crucial structural proteins, such as CTCF and RAD21, as well as the transcription machinery like RNA polymerase II (RNAPII) . The method provided base-pair resolution for a large number of interactions globally .
However, the traditional ChIA-PET protocol faced significant experimental hurdles.
- High Input Requirement: The original method demanded substantial cellular input, often exceeding $10^7$ cells, limiting its use with precious clinical or rare cell line samples.1
- Ex Situ Noise: Interactions were ligated outside the nucleus (ex situ), which increased the potential for non-native contacts and higher background noise.1
To address these limitations, protocols were refined, notably with the introduction of Advanced ChIA-PET. This version simplified the library process by incorporating the highly efficient Tn5 transposase for adapter ligation and fragmentation in a single step . This improvement laid the technical groundwork for the next generation.
HiChIP and PLAC-seq: Next-Generation Efficiency
HiChIP (High-Efficiency Chromatin Immunoprecipitation and Interaction Capture) represents a pivotal technical leap. It overcomes the limitations of ChIA-PET by incorporating innovations from Hi-C, dramatically boosting sensitivity and reducing required input.1
Core Technical Advantages of HiChIP
- In Situ Ligation: Chromatin contacts are chemically fixed and ligated inside the intact cell nucleus before cell lysis.1 This essential step prevents the formation of random, false-positive interactions, resulting in a much superior signal-to-background ratio compared to ex situ methods.1
- Minimal Input Volume: The high efficiency of the HiChIP workflow reduces the required cellular input by over 100-fold compared to traditional ChIA-PET.1 This sensitivity (often requiring $\le 10^5$ cells) makes HiChIP the standard choice for scarce samples.
- Increased Informative Yield: The optimized protocol improves the yield of conformation-informative reads by more than 10-fold.1 This ensures that sequencing effort is focused on genuine regulatory contacts, improving data quality and depth.
PLAC-seq: Specialized Regulatory Mapping
PLAC-seq, or Promoter Ligation Assisted Capture sequencing, is typically implemented as an optimized, specialized application of the HiChIP protocol. It is specifically standardized for mapping chromatin loops associated with well-defined functional histone marks.
Key Applications
PLAC-seq is particularly powerful for research into gene regulatory dynamics, focusing primarily on active promoters, typically targeted via H3K4me3, and active enhancers, defined by H3K27ac. By zeroing in on these key epigenetic signatures, PLAC-seq efficiently resolves functional promoter-enhancer loops genome-wide.
The data derived from PLAC-seq is highly valuable for integrative studies. For instance, high-resolution H3K27ac and H3K4me3 loop maps can be overlaid with disease risk variants identified through Genome-Wide Association Studies (GWAS). This integration helps researchers connect non-coding risk polymorphisms to their target genes, enabling the prioritization of novel genes related to disease pathogenesis. This provides a definitive mechanistic understanding of how genetic variation contributes to complex conditions, such as those related to aberrant enhancer regulation seen in autoimmune diseases.
Streamlining the Workflow
The design of HiChIP and PLAC-seq promotes exceptional efficiency in the laboratory. By combining and streamlining multiple steps into a single, efficient on-bead enzymatic reaction, the entire library construction process for HiChIP or PLAC-seq can often be reliably completed within a 2-day workflow.
Detailed Comparative Analysis: Optimizing Your Chromatin Interaction Strategy
Selecting the optimal Chromosome Conformation Capture method is vital for project success, balancing input constraints against the desired resolution and specificity. HiChIP and PLAC-seq offer significant technical advantages over the foundational ChIA-PET method, making them superior choices for most modern research applications.
Key Differentiators: A Technical Comparison
Table 1: Technical Comparison of Protein-Centric Chromatin Capture Technologies
| Feature | ChIA-PET (Traditional) | HiChIP | PLAC-seq |
| Core Workflow | ChIP first to enrich target protein-bound fragments, followed by Chromatin Interaction Linking. | In situ crosslinking/ligation first, followed by ChIP, then Tn5 enzyme-based library construction. | Similar to HiChIP: Chromatin Interaction Linking before fragmentation, with library preparation via end repair, A-tailing, and adapter ligation. 8 |
| Input Cells Required | High ($\ge 10^7$ cells) 9 | Low ($\le 10^5$ cells) 4 | Low ($\le 0.5 \text{M}$ cells) 10 |
| Sensitivity | Moderate (3–12% unique PETs) 9 | High (>40% unique PETs; $>\!10$-fold ChIA-PET) 9 | Moderate/High (Up to $100\times$ more cost-effective than ChIA-PET) 10 |
| Primary Advantage | High resolution; specific enrichment of target protein-mediated chromatin interactions; good for comprehensive analysis. | Lower cost and high sensitivity, making it compatible with low cell input and large-scale factor analysis (especially TFs/chromatin modifiers). | High specificity and good reproducibility for detecting tight spatial interactions (e.g., enhancer-promoter loops). |
| Key Limitations | Requires high sequencing depth and input material, leading to high cost and technical difficulty. | Potential preference for open chromatin regions; requires rigorous antibody optimization for efficiency. | Sensitive to MNase digestion conditions, potentially leading to loss of some long-range interaction information. |
| Optimal Application | Research requiring comprehensive, factor-mediated analysis in systems with abundant input material. | Functional studies of transcription factors and chromatin modification factors, especially with low cell input. | Analyzing fine regulatory interactions, particularly promoter-enhancer loops and distal DNaseI hypersensitive sites. |
Service Selection Guidance for Researchers
The choice between services should align precisely with the research question and sample availability.
- For Rare or Clinical Samples: We recommend HiChIP as the standard platform. Its $\le 10^5$ cell input requirement is critical for working with limited patient biopsies, sorted cell populations, or challenging primary cells. HiChIP is ideal for mapping structural proteins like Cohesin or CTCF, providing robust data on genome folding.1
- Internal Link Goal: [hichip-sequencing.html]
- For Gene Regulatory Focus: PLAC-seq is the specialized, optimal choice. By targeting active marks like H3K4me3 or H3K27ac, it yields precise, high-confidence maps of functional Promoter-Enhancer Chromatin Loops.2 This data is foundational for integration with genetic studies, such as GWAS, to link non-coding variants to their target genes and disease pathogenesis.2
- For Comprehensive Structure Mapping: Hi-C remains the indispensable tool for truly genome-wide analysis of the 3D Genome, necessary for identifying TAD boundaries and overall chromosomal organization, regardless of specific protein involvement.3
- Internal Link Goal: [hifi-c-sequencing.html]
Our protocols, whether employing HiChIP or PLAC-seq, are optimized for efficient capture of both 3D looping and the associated 1D ChIP enrichment data, ensuring a rapid and reproducible workflow.
Functional Applications in Research and Disease Modeling
Protein-centric 3D genomic mapping methodologies provide the mechanistic link between regulatory elements and their distant target genes, offering critical clarity for complex biological questions.
Dissecting Gene Regulatory Landscapes
The systematic utilization of HiChIP/PLAC-seq targeting H3K27ac and H3K4me3 allows for genome-wide mapping of active regulatory loops. This capability enables researchers to analyze the dynamics of these interactions during critical cellular state transitions, such as pluripotency and differentiation.
A powerful application involves the functional integration of this 3D data with large-scale genetic association studies. By combining high-resolution loop maps with disease risk variants, researchers can transition from simple association to mechanistic understanding, prioritizing and validating novel genes whose regulatory input is affected by genetic variants, thereby informing disease pathogenesis.
Mapping Structural Architecture and Dynamics
HiChIP is indispensable for studying the larger-scale, long-range genome folding dictated by architectural proteins like CTCF and Cohesin. By capturing these structural elements, researchers can effectively explore the impact of mis-folding in diseases such as cancer or understand the structural basis underlying normal promoter-enhancer communication. The ability to accurately map these dynamics is fundamental to designing interventions that modulate specific pathological chromatin structures.
Advanced Bioinformatics for 3D Genomic Data Integrity
The output of any protein-centric 3D genomic assay is only as reliable as the bioinformatics pipeline used for processing. Expertise in next generation sequencing bioinformatics is crucial for navigating the inherent complexity of chimeric reads and accurately modeling background noise.
Statistical Modeling for Accurate Contact Frequency Determination
The data confirms that the structural information derived from Chromatin Interaction Linking—the 3D contact frequency—is a more prominent determinant of interaction accuracy than the 1D presence of the binding factor. Therefore, advanced statistical rigor is required to accurately model the relationship between the 1D enrichment and the 3D contacts.
Raw contact frequencies are affected by systematic biases, including differences in regional ChIP enrichment. To achieve reliable, normalized contact maps, specialized computational pipelines must implement advanced normalization methodologies.
The deployment of Positive Poisson Regression (PPR) models is mandatory for rigorous analysis. This sophisticated framework is utilized for several critical steps:
- Normalization and Bias Correction: PPR models are used to fit the data and normalize for the HiChIP/PLAC-seq specific ChIP enrichment (the 1D signal). The resulting residuals provide the normalized contact frequencies, effectively correcting for systematic biases and ensuring that reported interactions are not simply artifacts of local enrichment.
- Noise Estimation: This methodology is applied to accurately estimate the noise level, which is particularly vital for correctly assessing the significance of contacts, especially for inter-chromosomal pairs.
- Differential Analysis: Robust pipelines analyze bin pairs (the anchors of the putative loops) based on their enrichment profiles (e.g., AND sets where both anchors are enriched versus XOR sets where only one is enriched). This nuanced analysis ensures that the differing ChIP enrichment levels at the two interacting anchors are properly accounted for during contact detection.
Quality Control and Reproducibility Standards
The final data delivery adheres to the highest standards of data integrity and reproducibility. Rigorous quality control metrics are applied to ensure consistency across biological replicates, demonstrating low variability between chromosomes, consistent with established Hi-C analysis standards (HiCRep). This commitment to computational transparency and statistical rigor ensures that the final delivery comprises high-confidence loop calls, normalized contact matrices, and corresponding 1D ChIP-enrichment profiles suitable for publication and functional validation.
Key Bioinformatics Considerations for Protein-Centric 3D Data
Table 2 highlights the essential computational steps necessary to convert raw data into a reliable, functional map of 3D genomic interactions, underscoring the necessity of expert bioinformatics analysis.
Table 2: Key Bioinformatics Considerations for Protein-Centric 3D Data
| Analytical Challenge | Required Methodology/Tool Principle | Technical Justification |
| Interaction Artifact Reduction | Inherent use of in situ data; Chimeric Read Utilization 3 | Minimizes ex situ artifacts and maximizes signal from paired tags, essential for robust loop calling. |
| Statistical Normalization | Positive Poisson Regression Models (PPR) 5 | Accurately models and corrects for ChIP enrichment bias and stochastic noise distribution, providing normalized contact frequencies. |
| Loop Calling | Specialized algorithms for identifying statistically significant contacts | Differentiates true regulatory interactions (loops) from random collision events and proximity artifacts. |
| Visualization & Interpretation | Genome Browser tracks (1D) and Circos/Heatmaps (3D) | Enables researchers to rapidly integrate 1D binding data with 3D looping structure for functional validation. |
Conclusion and Recommendations
The technological evolution to HiChIP and PLAC-seq has decisively addressed the input and sensitivity limitations of the initial ChIA-PET methodology. By leveraging the principles of in situ ligation and on-bead Tn5 tagmentation, these modern methods provide an efficient and high-fidelity approach to protein-centric 3D genome mapping.
For research demanding the highest sensitivity and lowest input, HiChIP is recommended for architectural proteins (CTCF, Cohesin), and PLAC-seq is recommended for promoter-enhancer interactions defined by active histone marks (H3K4me3, H3K27ac).
Crucially, the reliability of the resulting 3D data is inseparable from the rigor of the next generation sequencing bioinformatics analysis. The deployment of advanced statistical models, such as Positive Poisson Regression, is mandatory to correct for inherent biases and confirm the statistical significance of identified chromatin loops. By combining superior molecular protocols with expert bioinformatics, the service provides authoritative 3D genome data essential for dissecting functional regulatory mechanisms in complex biological systems.
Selecting Your 3D Genome Strategy and Next Steps
Deciding on the right Chromosome Conformation Capture method—whether global or protein-centric—is the most critical initial project decision. Our specialized services are structured to provide authoritative data for any 3D Genome question, backed by robust protocols and rigorous next generation sequencing bioinformatics.
Service Selection Summary
We offer a complete suite of services tailored to your research focus:
Goal Recommended Service Input Required Focus
Comprehensive 3D Structure Hi-C / Hi-C Sequencing Medium to High Unbiased, global view (TADs, large-scale Chromatin Loop structures)
Protein/Factor Mechanism HiChIP Sequencing Very Low ($\le 10^5$ cells) High-sensitivity mapping of structural factor interactions (e.g., Cohesin, CTCF) 1
Gene Regulation Focus PLAC-seq (H3K27ac/H3K4me3) Very Low ($\le 10^5$ cells) Functional Promoter-Enhancer Chromatin Loops and regulatory network analysis
Locus-Specific Analysis 4C-seq Medium All interactions connected to a single genomic viewpoint (e.g., specific promoter)
Commitment to Quality and Deliverables
Reliable interaction mapping requires more than just sequencing reads. Our commitment extends to delivering statistically robust, functionally relevant data packages:
- Validated Protocols: Use of in situ ligation (for HiChIP and PLAC-seq) minimizes non-native interactions, guaranteeing high fidelity.1
- Expert Bioanalysis: Deployment of the Positive Poisson Regression (PPR) model for accurate normalization and high-confidence loop calling, essential for publishing high-impact findings .
Start Your 3D Genome Project
Ready to move beyond 1D mapping and unlock the complex regulatory mechanisms of the 3D Genome? Our expert team can help design the optimal Chromatin Interaction capture strategy based on your unique sample and research goals.
Contact us today to start your high-resolution HiChIP or PLAC-seq project.
Frequently Asked Questions (FAQs)
What is the primary advantage of HiChIP and PLAC-seq over traditional ChIA-PET for Chromatin Interaction analysis?
The key advantage is unparalleled efficiency and low input requirement. HiChIP and PLAC-seq significantly enhance efficiency by performing Chromatin Interaction Linking in situ (within the nucleus) prior to cell lysis, thereby minimizing false-positive interactions. Quantitatively, HiChIP improves the yield of conformation-informative reads by over 10-fold and reduces the cell input requirement by over 100-fold compared to ChIA-PET, which typically requires hundreds of millions of cells. Similarly, PLAC-seq is nearly 100 times more cost-effective than ChIA-PET, even when using 20-fold fewer cells.
Which specific regulatory elements are best mapped using PLAC-seq?
PLAC-seq is the optimal technique for high-resolution mapping of active regulatory elements, with a primary focus on Promoter-Enhancer Chromatin Loops. This is achieved by targeting active histone marks such as H3K4me3 (active promoters) and H3K27ac (active enhancers). The resulting high-resolution loop maps can be integrated with external genetic data (like GWAS) to prioritize novel disease-related genes.
How do protein-centric methods (HiChIP/PLAC-seq) differ from global methods like Hi-C?
Hi-C is designed to map all chromatin interactions across the entire genome at an accurate genome-wide scale, providing a map of the overall 3D structure. In contrast, protein-centric methods (HiChIP and ChIA-PET) incorporate an immunoprecipitation (ChIP) step to enrich for DNA fragments associated with a specific protein (e.g., CTCF or H3K27ac). This focuses the sequencing capacity solely on the interactions mediated by the factor of interest, resulting in higher specificity and a superior signal-to-noise ratio for mechanistic analysis.
How does your bioinformatics pipeline ensure the reliability of the detected chromatin loops?
The reliability of 3D contact data is ensured through sophisticated statistical analysis. Our pipeline utilizes the Positive Poisson Regression (PPR) model. This model accurately normalizes the data to correct for ChIP enrichment bias (the 1D signal) and other systematic biases, using the residuals as the actual, normalized contact frequencies. This rigorous normalization process is crucial for differentiating true functional loops from random proximity events, ensuring high-confidence loop calls.
Can HiChIP and PLAC-seq be used for rare cells or clinical samples?
Yes, this is the primary strength of these next-generation techniques. HiChIP's efficient in situ protocol and use of Tn5 tagmentation dramatically reduce the required cell input, making it compatible with rare cell populations, primary cells, and human tissue samples—systems that were often unmeasurable using conventional ChIA-PET strategies. For example, strong long-range interactions have been detected by PLAC-seq using as few as 0.5 million cells.
References
- Wang Y, Song F, Zhang B, Zhang L, Xu J, Kuang D, Li D, Choudhary MNK, Li Y, Hu M, Hardison R, Wang T, Yue F. The 3D Genome Browser: a web-based browser for visualizing 3D genome organization and long-range chromatin interactions. Genome Biol. 2018
- Reyna J, Fetter K, Ignacio R, Ali Marandi CC, Ma A, Rao N, Jiang Z, Figueroa DS, Bhattacharyya S, Ay F. Loop Catalog: a comprehensive HiChIP database of human and mouse samples. bioRxiv [Preprint]. 2025
- Fullwood M J, Liu M H, Pan Y F, et al. An oestrogen-receptor-α-bound human chromatin interactome. Nature, 2009, 462(7269): 58-64.
- Li X, Luo O J, Wang P, et al. Long-read ChIA-PET for base-pair-resolution mapping of haplotype-specific chromatin interactions. Nature protocols, 2017, 12(5): 899-915.
- Wang P, Feng Y, Zhu K, et al. In Situ Chromatin Interaction Analysis Using Paired‐End Tag Sequencing. Current protocols, 2021, 1(8): e174.
- Mumbach M R, Rubin A J, Flynn R A, et al. HiChIP: efficient and sensitive analysis of protein-directed genome architecture. Nature methods, 2016, 13(11): 919-922.
- Zeng W, Liu Q, Yin Q, et al. HiChIPdb: a comprehensive database of HiChIP regulatory interactions. Nucleic acids research, 2023, 51(D1): D159-D166.
- Fang R, Yu M, Li G, et al. Mapping of long-range chromatin interactions by proximity ligation-assisted ChIP-seq. Cell research, 2016, 26(12): 1345-1348.
- Yu M, Zemke N R, Chen Z, et al. Integrative analysis of the 3D genome and epigenome in mouse embryonic tissues. Nature Structural & Molecular Biology, 2024: 1-12.
- Handoko L, Xu H, Li G, et al. CTCF-mediated functional chromatin interactome in pluripotent cells. Nature genetics, 2011, 43(7): 630-638.
For research purposes only, not intended for personal diagnosis, clinical testing, or health assessment