ONT Direct RNA Sequencing: From Real-Time Detection to Analytical Challenges
The development of RNA sequencing technology has been driven by two fundamental objectives: the ability to accurately analyze the entire transcriptome and the capacity to dynamically capture RNA modification information. Conventional short-read-long sequencing technologies encounter limitations due to fragmented splicing, impeding the accurate identification of full-length transcripts, complex shear variants, and fusion genes. In contrast, antibody-based modification detection methods face constraints related to low throughput and high false-positive rates. In light of these limitations, Oxford Nanopore direct RNA sequencing technology has emerged as a significant development. This technology subverts the conventional sequencing process by detecting natural RNA molecules through nanopore real-time electrical signals, facilitating full-length transcript analysis without the need for reverse transcription or amplification, and preserving RNA modification features simultaneously. This technology not only overcomes the limitations of short-read-long technology in analyzing complex shear sites but also provides a new tool for epitope transcription regulation research through multi-dimensional data integration.
This article systematically delineates the core principles and technical advantages of ONT direct RNA sequencing. The article discusses how current changes induced by RNA molecules are captured by nanopores in real time. It explains how this process can directly analyze the complete transcript structure and identify modification sites, avoiding the amplification bias of traditional methods. The article goes on to describe how its long read-length feature can accurately analyze complex shear sites, fusion genes, and poly-tailed heterogeneity. This, the article asserts, can significantly enhance the sensitivity of low-abundance transcript detection. The article also discusses optimization strategies and data analysis tools. Additionally, it identifies challenges to clinical translation, including error rate control, massive data management efficiency, and a lack of cross-platform standardization. Finally, by contemplating prospective applications of quantum computing, multi-omics integration, and artificial intelligence-assisted diagnosis, the article underscores the pivotal function of technological iteration and interdisciplinary collaboration in propelling RNA modification research from fundamental to clinical domains.
Service you may intersted in
Learn More:
Principles of ONT Direct RNA Sequencing
ONT direct RNA sequencing technology has fundamentally transformed the landscape of transcriptome research. This technology has achieved a breakthrough in real-time detection and full-length transcript resolution without the need for amplification, which represents a significant advancement in the field. In contrast to conventional RNA sequencing methods, which depend on reverse transcription, fragmentation, and short-read splicing, this technology employs a direct approach by reading natural RNA molecules through nanopores. This not only preserves the original modification information of RNA but also accurately captures the complete structure of the transcriptome across complex shear sites. The subsequent analysis will examine the real-time detection mechanism and the advantages of long read length technology.
Real-Time RNA Detection via Nanopores
The core of ONT direct RNA sequencing technology is the real-time capture of the current changes triggered by single-stranded RNA molecules as they travel through the nanopore. When RNA is driven through the nanopore by an electric field, the disparities in the chemical structure and charge distribution of different nucleotides or modified bases exert a particular blocking effect on the ion flow. This transient current signal is then recorded by a high-precision sensor and subsequently decoded into base sequence information by a machine learning algorithm. For instance, RNA modifications such as pseudouridine (Ψ) and m6A have been shown to alter the local conformation or charge, thereby generating current features that differ significantly from unmodified bases. Consequently, these modifications can be recognized directly. This technology eliminates the reverse transcription or PCR amplification step in conventional sequencing, enabling direct sequencing of natural RNA molecules. This approach circumvents amplification bias errors, such as GC bias, while preserving the original modification information of the RNA. The researchers were able to locate the modification sites with high precision by analyzing the perturbation of ion flow trajectories. Furthermore, the incorporation of a real-time analysis feature enables the dynamic adjustment of experimental strategies during the process of sequencing. This adjustment, for instance, facilitates the rapid targeting of key sequences in the context of pathogen surveillance. Consequently, this facilitates the provision of immediate support for precision medicine.
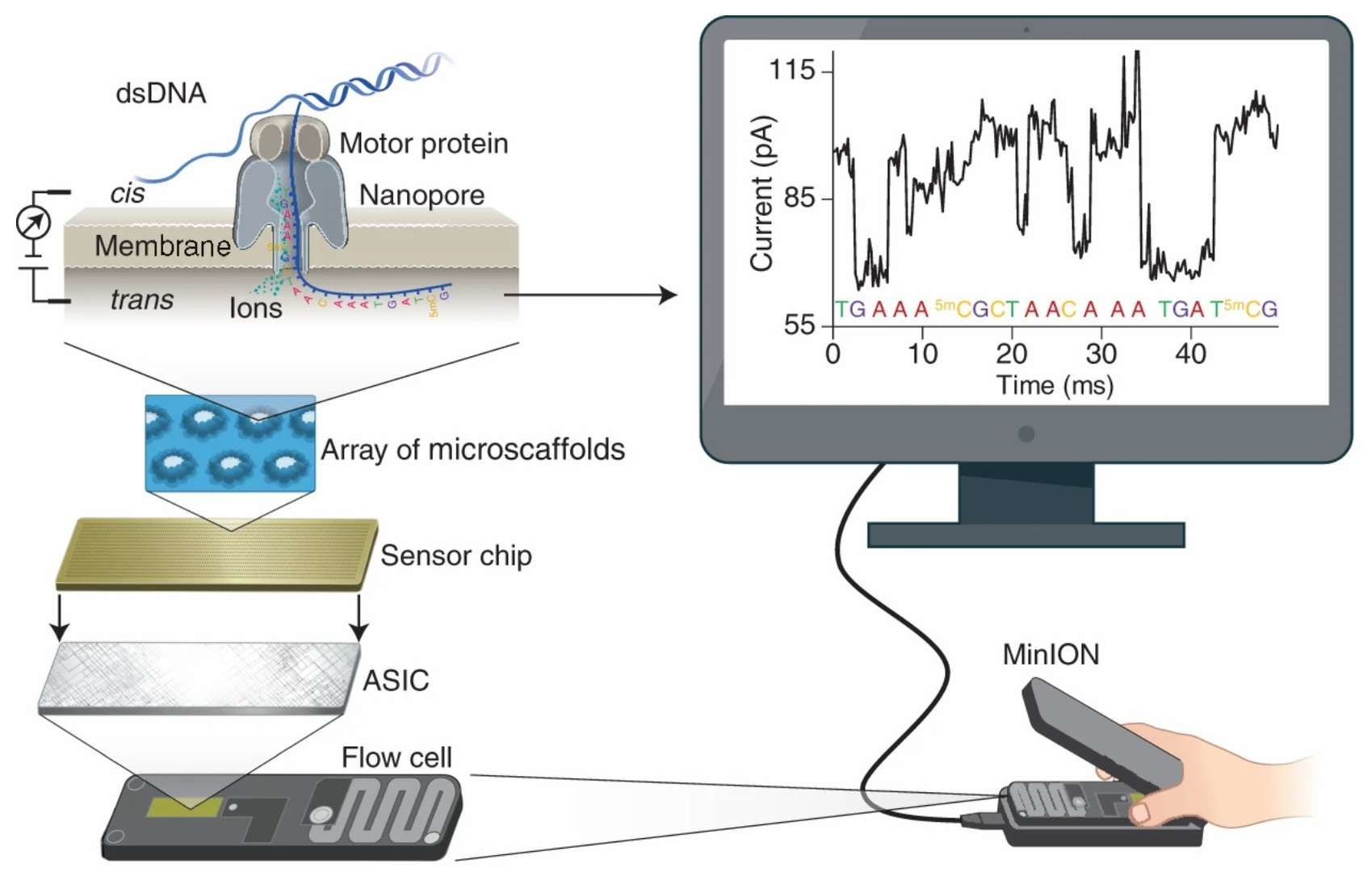 Principle of nanopore sequencing (Wang et al., 2021)
Principle of nanopore sequencing (Wang et al., 2021)
Full-Length Transcript Analysis vs. Short-Read Sequencing
ONT direct RNA sequencing has demonstrated notable advantages in full-length transcript resolution due to its long read length characteristics (average >1 kb, covering the full transcript length). Conversely, short read length sequencing technologies (e.g., Illumina) employ fragment splicing to infer transcript structure due to read length limitations (typically ≤300 bp). This approach often results in misclassification of splice variants and failure to detect fusion genes. For instance, complex shear patterns (e.g., nested exons) are frequently overlooked due to the limitations of short read lengths, which cannot span multiple junctions. Additionally, low-coverage regions near fusion gene breakpoints are susceptible to misclassification as sequencing errors. Conversely, ONT has been demonstrated to directly capture intact RNA molecules and accurately resolve variable shear sites, fusion gene breakpoints, and poly(A) tail heterogeneity. The study demonstrates that the utilization of Nanopore data-based analysis tools enables the detection of low-frequency fusion isoforms, which conventional short-read-length techniques are often unable to recognize, including the rare fusion events reported in certain cancer studies. Moreover, ONT's capacity to function without PCR amplification obviates the influence of GC preference on quantitative analysis, thereby markedly enhancing the sensitivity of detection of low-abundance transcripts and rare isoforms. Furthermore, its single sequencing can synchronously integrate multi-dimensional information, including original RNA sequence, modification sites (e.g., m6A, Ψ), and structural features (e.g., poly(A) tail length). This provides a new perspective for analyzing the dynamic interactions of RNA modifications (e.g., the mutually exclusive effects of m6A and pseudouridine in translation).
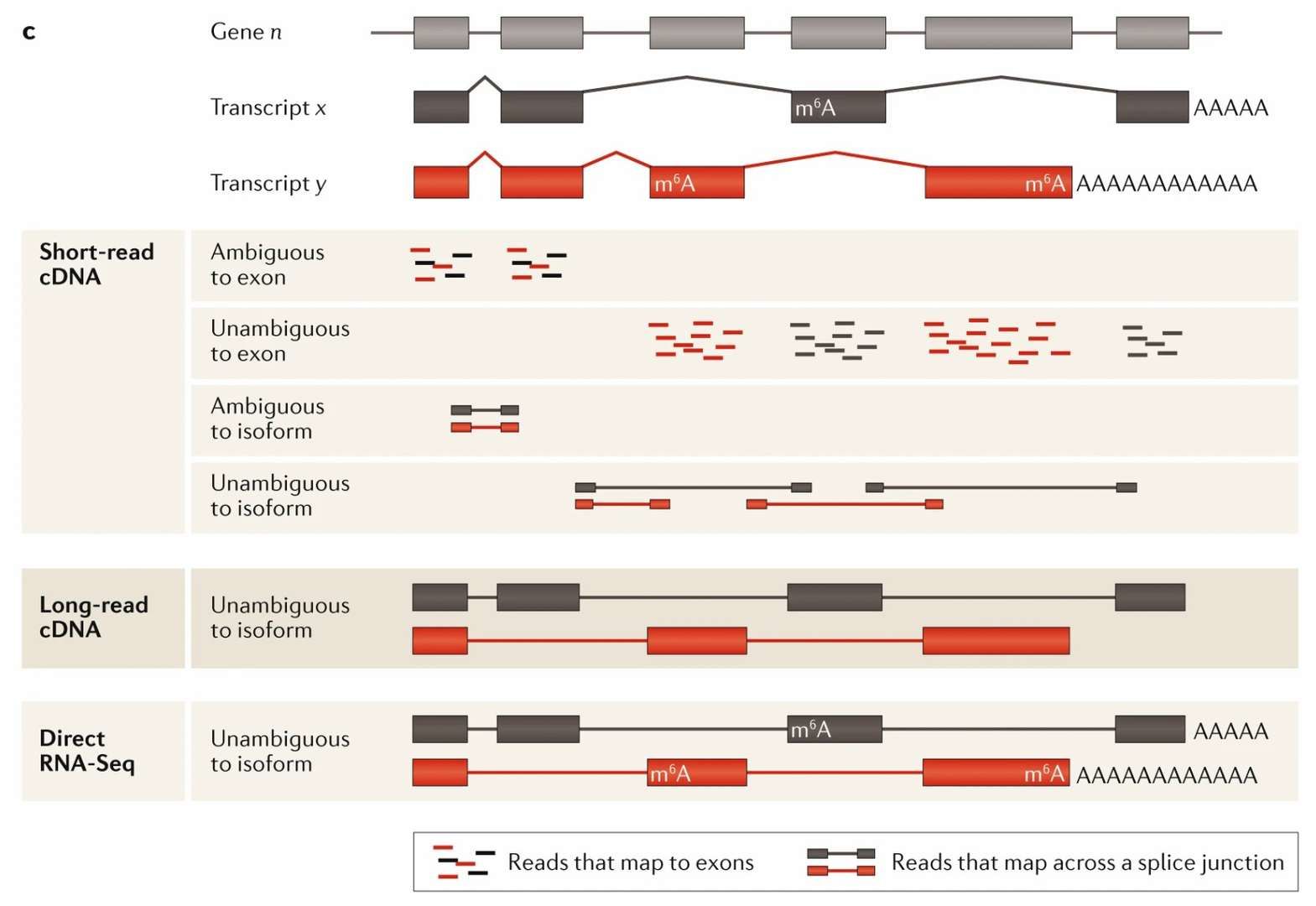 Comparison of short-read, long-read and direct RNA-seq analysis (Stark et al., 2019)
Comparison of short-read, long-read and direct RNA-seq analysis (Stark et al., 2019)
Workflow Optimization Strategies
The process optimization of Oxford Nanopore direct RNA sequencing is focused on two core aspects: the efficient construction of RNA libraries and the stability control of real-time data generation. Through the implementation of rigorous quality control measures to ensure RNA integrity, the optimization of nanopore threading orientation, and the integration of algorithms and hardware co-design, the technology has been shown to enhance the accuracy and applicability of long read length sequencing.
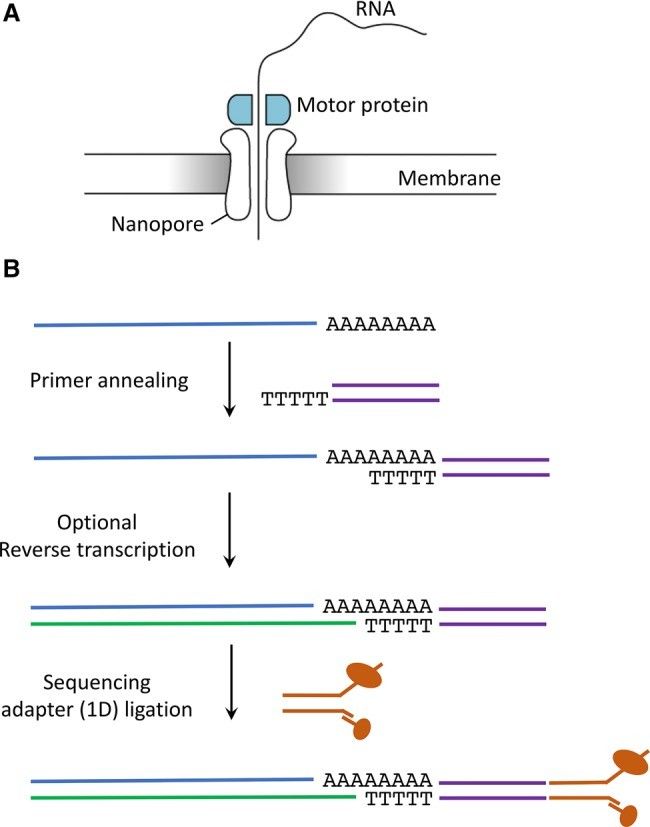 Direct RNA sequencing library preparation steps using Oxford Nanopore Technologies (Jonkhout et al., 2017)
Direct RNA sequencing library preparation steps using Oxford Nanopore Technologies (Jonkhout et al., 2017)
RNA Preparation and Adapter Ligation
Oxford Nanopore's SQK-RNA002 kit has been demonstrated to enhance the efficiency of library construction for direct RNA sequencing by implementing rigorous quality control measures and optimizing procedural steps. The RNA must meet a minimum RNA integrity number (RIN) of 8 to ensure the integrity of the library preparation. The high-quality RNA is essential to avoid 3' end preference for sequencing coverage and to reduce the risk of junction ligation failures or elevated proportions of repetitive sequences. The SQK-RNA002 kit utilizes the poly(A) tail anchoring mechanism to guide RNA molecules through the nanopore in the 5'→3' direction. The poly(A) tail's α-helical structure prolongs nanopore blocking time and enhances signaling stability. For non-poly(A) samples, such as viral RNA, the kit extends the range of applicability of the technology by enzymatically adding artificial poly(A) tails. Furthermore, the library construction process integrates the removal of ribosomal RNA, multiplexing adapter pre-ligation, and efficient reverse transcription in a mere 2 hours without fragmentation, thereby preserving full-length transcript information. This characteristic renders the process particularly well-suited for the analysis of low-abundance RNA.
Real-Time Data Generation and Flow Cell Stability
Real-time data generation is contingent upon the implementation of deep learning and hardware acceleration techniques within the Guppy software framework. The software utilizes recurrent neural networks (RNNs) to parse nanopore electrical signals, supports single-stranded and double-stranded sequencing modes, and flexibly switches between fast mode (~500 reads/s) and high-precision mode (Q ≥ 7) to meet the demands of clinical real-time monitoring and scientific research with high precision. Ensuring data continuity is paramount, and the Guppy software is designed to minimize voltage fluctuations and bubble interference through constant current mode, a serpentine flow channel structure, and active pore maintenance (e.g., regular cleaning and pre-experiment QC) to ensure electrical signal consistency. Environmental factors such as temperature fluctuation (within ±1°C) and ion concentration stability (e.g., 1M KCl buffer system) have been demonstrated to directly affect sequencing efficiency. When utilized in conjunction with Guppy's real-time base identification and flow cell optimization technologies, this approach has been demonstrated to achieve transcript coverage with a median read length of 771 bp and a maximum read length of over 21 kb. Furthermore, it facilitates dynamic target sequence screening, including mitochondrial RNA enrichment, which has been shown to markedly enhance the capacity to resolve complex isoforms.
Data Analysis for Accuracy and Insight
The in-depth analysis of Oxford Nanopore sequencing data relies on the synergistic analysis of error correction and transcriptome visualization. Through the integration of signal optimization algorithms, machine learning models, and multi-dimensional visualization tools, researchers can extract high-precision sequence information from raw electrical signals. This process enables the identification of post-transcriptional modifications and variable shear events, leading to the revelation of disease-related molecular regulatory networks. The subsequent discussion will address the pivotal function of the data analysis process in enhancing the biological interpretation of sequencing outcomes, with particular emphasis on error correction tools and transcriptome visualization techniques.
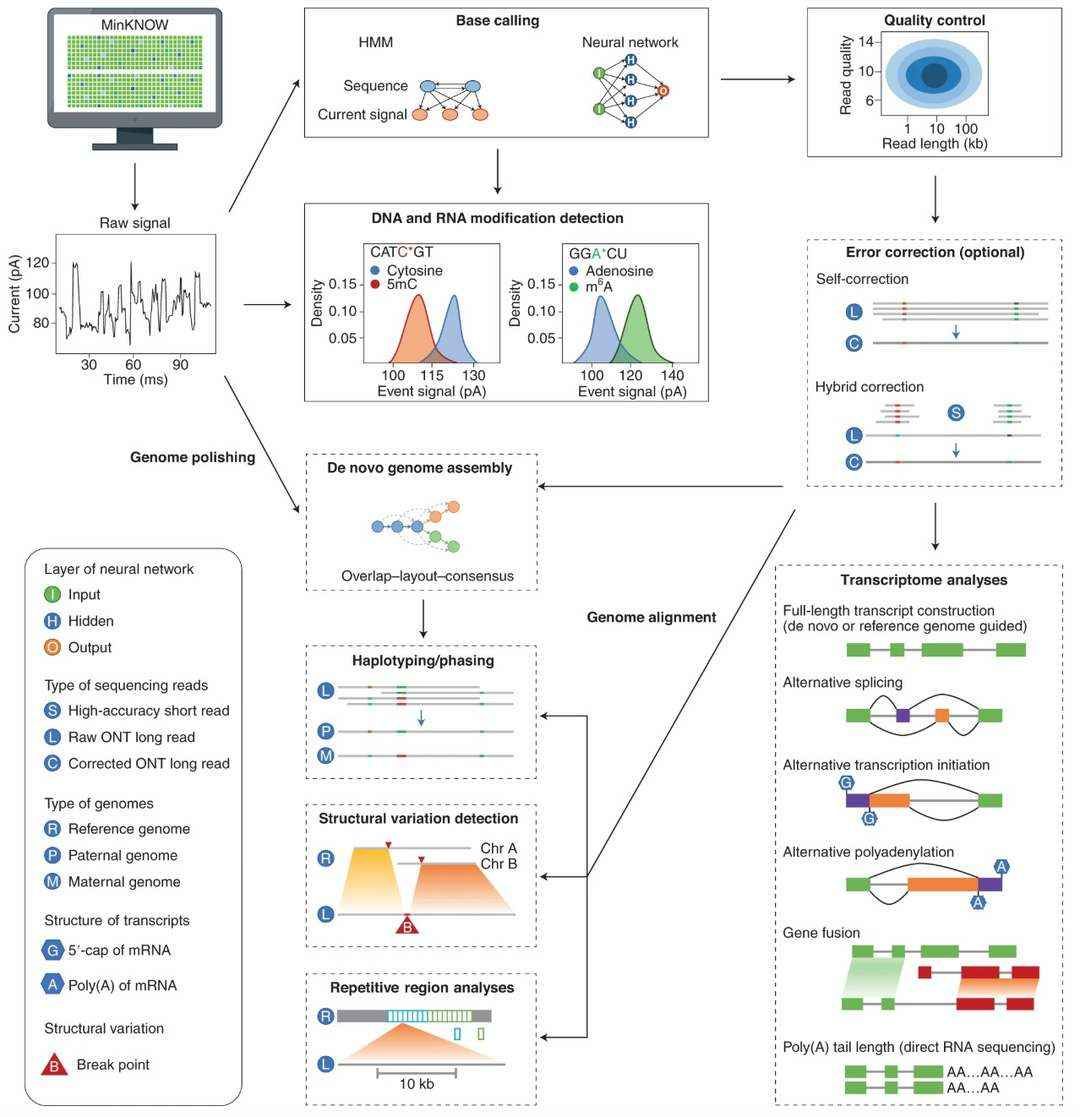 Analyses of ONT sequencing data (Wang et al., 2021)
Analyses of ONT sequencing data (Wang et al., 2021)
Error Correction Tools: Nanopolish, Medaka, and EpiNano
The Nanopolish software utilizes Hidden Markov Models to optimize raw signals, generate high-precision consensus sequences by comparing long read-length data with reference genomes, and facilitate methylation site detection and variant analysis of low-coverage regions. For instance, in the context of bacterial genome assembly, it has been observed to substantially correct methylation-related errors and enhance processing efficiency through the utilization of parallel computing. Concurrently, the complementary Medaka employs neural network modeling to markedly reduce assembly error rates under specific sequencing conditions and operates at a substantially higher velocity than conventional tools. In the context of RNA epigenetic modifications, EpiNano utilizes machine learning algorithms to systematically analyze error characteristics, thereby achieving a control group-independent m6A detection capability. This approach has been demonstrated to possess unique application value in clinical samples, as evidenced by its success in identifying RNA modification sites associated with metabolic pathways in plant pathogen research.
Transcriptome Visualization: StringTie and IGV Browser
The ability to effectively address the intricacies of the transcriptome is contingent upon the utilization of sophisticated assembly and visualization tools. StringTie, a notable example, employs optimized algorithms to reconstruct full-length transcripts, identify a substantial number of variable shear isoforms in human samples, and integrate quantitative expression data to support functional studies. For instance, it has been demonstrated that StringTie can reveal the association of non-coding RNAs with regulatory pathways in studies of model organism aging. In contrast, the Integrative Genomics Viewer (IGV) facilitates the dynamic display of multidimensional data, ranging from a whole-genome perspective to single-base resolution for precise localization. In the context of cancer research, IGV has been instrumental in elucidating the co-occurrence of disease-associated splicing variants and chromatin modification hotspots by overlaying epigenomic modification and transcriptomic data, thereby providing intuitive evidence for mechanistic research.
Challenges and Future Directions
The clinical application of RNA modification detection technology is still facing core challenges, such as error rate control, data management efficiency, and lack of standardization, despite the rapid development of the technology. The present study aims to address these challenges by exploring methodologies for reducing false-positive interference while enhancing detection sensitivity. Additionally, it seeks to investigate approaches that facilitate cross-platform data comparability through open-source collaboration and technological innovation. The subsequent discussion explores the pivotal roles of technological innovation and community collaboration in propelling future advancements, with a focus on error rate reduction and data management optimization, as well as the establishment of clinical standards.
Error Rate and Data Management
The accuracy of RNA modification detection is constrained by base recognition bias in relation to the efficiency of processing voluminous data sets. For instance, the misclassification problem of m6A modifications in nanopore sequencing and the nonspecific binding of antibody-based methods (e.g., MeRIP-seq) can result in false positive signals. To address these challenges, researchers have proposed to enhance the reliability of results by improving base recognition models and optimizing data management strategies. The employment of base recognition models based on unmodified RNA training, such as the IVT model, has been demonstrated to enhance the sensitivity of modification site detection. Moreover, the integration of synthetic unmodified RNA libraries provides a standardized negative control for experiments, thereby effectively mitigating background noise. In terms of data management, the synergistic application of edge computing and cloud platforms becomes pivotal in reducing transmission delays through real-time preprocessing and distributed computing. This approach is further enhanced by dynamic resource scheduling strategies, which optimize analysis efficiency. The development of modification-specific recognition algorithms and the exploration of quantum computing in pattern recognition are expected to further break through the limitations of existing technologies.
Clinical Standardization
A significant impediment to the clinical translation of these technologies is the absence of methodological differences and the inability to perform meaningful data comparisons. Assay results from different technology platforms (e.g., nanopore sequencing vs. mass spectrometry) can vary significantly, and the uncertainty in results is further exacerbated by the fluctuating reproducibility of antibody-dependent experiments. In light of these challenges, the role of open-source tools and community collaboration in promoting standardization has become increasingly salient. For instance, the DRUMMER tool, which is based on nanopore data, facilitates single-nucleotide resolution detection through statistical testing with background noise correction, while xPore supports cross-sample differential modification analysis, thereby providing a unified framework for multicenter studies. Furthermore, the establishment of public databases and the promotion of gold standards (e.g., IVT RNA libraries) have led to substantial advancements in the integration of cross-platform data. In the future, the integration of standardized kits, the enhancement of the regulatory framework, and the combination of multi-omics data with artificial intelligence-assisted diagnosis are expected to further expand the role of RNA modification testing in disease marker discovery and the development of individualized medicine.
References
- Stark, Rory et al. "RNA sequencing: the teenage years." Nature Reviews Genetics. 20 (2019): 631 - 656. doi:10.1038/s41576-019-0150-2
- Wang, Yunhao et al. "Nanopore sequencing technology, bioinformatics and applications." Nature biotechnology. 39,11 (2021): 1348-1365. doi:10.1038/s41587-021-01108-x
- Jonkhout, Nicky et al. "The RNA modification landscape in human disease." RNA. 23,12 (2017): 1754-1769. doi:10.1261/rna.063503.117






