Interpreting the Insights from Long Amplicon Sequencing in Plant Research
At a glance:
- Brief Introduction of Plant Long Amplicon Sequencing
- Case Study of Plant Long Amplicon Sequencing
- Challenges of Plant Long Amplicon Sequencing
- Conclusion
As the "second genome" of plants, plant microbiome has a far-reaching impact on plant biological activities. In terms of nutrient absorption, rhizosphere microorganisms, such as arbuscular mycorrhizal fungi, can form symbionts with plant roots, and transform nutrients such as phosphorus and zinc in soil that are difficult to be directly absorbed by plants into usable forms, which significantly improves the nutrient uptake efficiency of plants.
The traditional short reading amplicon sequencing technology is widely used in the research of plant microbiome because of its advantages of Qualcomm and low cost. However, its short reading length (usually 150-300bp) faces many challenges in analyzing complex microbial community structure. For species with high similarity, it is difficult for short reading sequences to provide enough specific information for accurate differentiation, which leads to incorrect annotation of species or inability to distinguish related species.
In addition, the function prediction based on short reading sequences often depends on the reference database. Due to the limitations of the database, it is difficult to accurately predict the functions of many unknown or rare microorganisms. However, the long amplicon sequencing technology can produce long reading sequences of thousands or even tens of thousands of base pairs. These long reading sequences can completely cover the full length of microbial genes such as 16S rDNA, 18S rDNA or ITS, and provide richer phylogenetic information, which is helpful to accurately identify high similarity species and analyze complex community structure.
This paper expounds the advantages of long amplicon sequencing technology in plant research, analyzes its application through related cases, discusses the challenges, and looks forward to its development prospect in the field of botany.
Brief Introduction of Plant Long Amplicon Sequencing
Long amplicon sequencing of plant is based on PCR technology. Specific primers are designed for target gene regions (such as prokaryotic 16S rDNA, eukaryotic 18S rDNA or ITS) to amplify sample DNA and enrich target DNA fragments. Subsequently, the direct reading of long DNA sequences was completed by single molecule sequencing technology.
Long-reading amplicon sequencing technology has brought a revolutionary breakthrough for botany research because of its long reading length, high-precision sequence analysis ability and efficient coverage of complex genome regions.
Long reading length: It can read longer DNA fragments, span a larger genome area compared with short reading sequencing technology, reduce splicing errors, analyze complex gene structure and variation more accurately, completely cover long intron regions or highly repetitive sequence regions in plant genes, and help to accurately identify the complete structure and potential regulatory elements of genes.
High accuracy: Long-read sequencing technology has high accuracy in base recognition and sequence splicing, and can detect genetic variations such as single nucleotide polymorphism (SNP) and insertion deletion (InDel) more accurately, which is of great significance for plant genotyping and genetic map construction.
Identification of complex variations: Complex structural variations, such as gene fusion, inversion and repetition, can be better found and analyzed, which may play a key role in plant evolution, adaptation and trait formation, and help to deeply understand the evolution and function of plant genome.
Case Study of Plant Long Amplicon Sequencing
In the field of botany research, the precise analysis of plant gene function, the in-depth exploration of species evolution relationship and the comprehensive evaluation of genetic diversity have always been the goals pursued by researchers. Combined with specific cases, the key role and remarkable achievements of plant long amplicon sequencing technology in practical application were deeply analyzed.
Reveal the Mechanism of Wheat Drought Resistance
Drought is one of the biggest obstacles to global agricultural productivity. Due to the influence of global climate change and human activities in this century, the frequency and severity of drought stress are increasing every year. Wheat (Triticum aestivum L.), as a staple food, accounts for more than 20% of global calorie intake. Although a variety of wheat varieties have been planted all over the world, the average annual output has decreased by 58-72% when they encounter drought stress in their growth stages. Therefore, improving the drought resistance of wheat is an urgent need to improve crop yield and global food security.
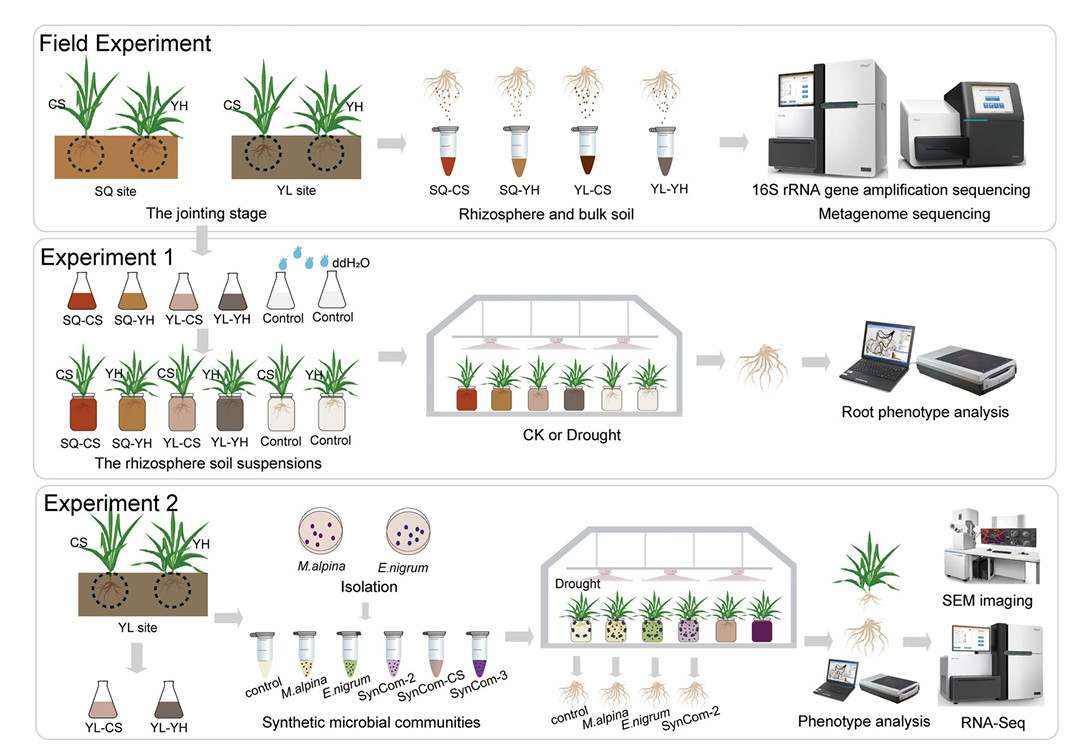 Experimental flow charts for this research (Yue et al., 2024)
Experimental flow charts for this research (Yue et al., 2024)
Firstly, the soil and rhizosphere microbial community diversity of drought-resistant (YH) and drought-sensitive (CS) varieties in SQ and YL planting sites were compared. Shannon index and Chao1 abundance of rhizosphere bacterial communities of YH varieties in SQ and YL locations were significantly higher than those of CS varieties. The variation of rhizosphere fungal communities in diversity and abundance showed a consistent pattern. The results showed that the diversity of rhizosphere microbial community of drought-resistant varieties was higher than that of drought-sensitive varieties, regardless of planting area.
For the variation of microbial community in YL planting site, PERMANOVA analysis further revealed that the biggest influence on soil microbial community was the compartment niche, followed by the plant genotype. Similarly, in SQ planting site, plant genotype can also explain the relatively high proportion of changes in the total microbial population. CAP and PCoA analysis also showed that host plant characteristics, compartment niche and planting location jointly affected the changes of soil bacterial and fungal communities. These results show that the bacteria and fungi groups of drought-resistant wheat are more diverse than those of drought-sensitive wheat.
These results show that different drought response types are closely related to plant genotypes. In addition, by analyzing the database of plant beneficial microorganisms and pathogenic microorganisms, the researchers identified 24 plant pathogenic fungi in all soil samples. Among these pathogenic fungi, the most important fungi are Ascomycetes (22 genera), followed by Basidiomycetes (1 genus), Cladosporium (1 genus) and Hyphantria (1 genus). Regardless of plant species, the relative abundance of ascomycetes at SQ locus was significantly higher than that at YL locus.
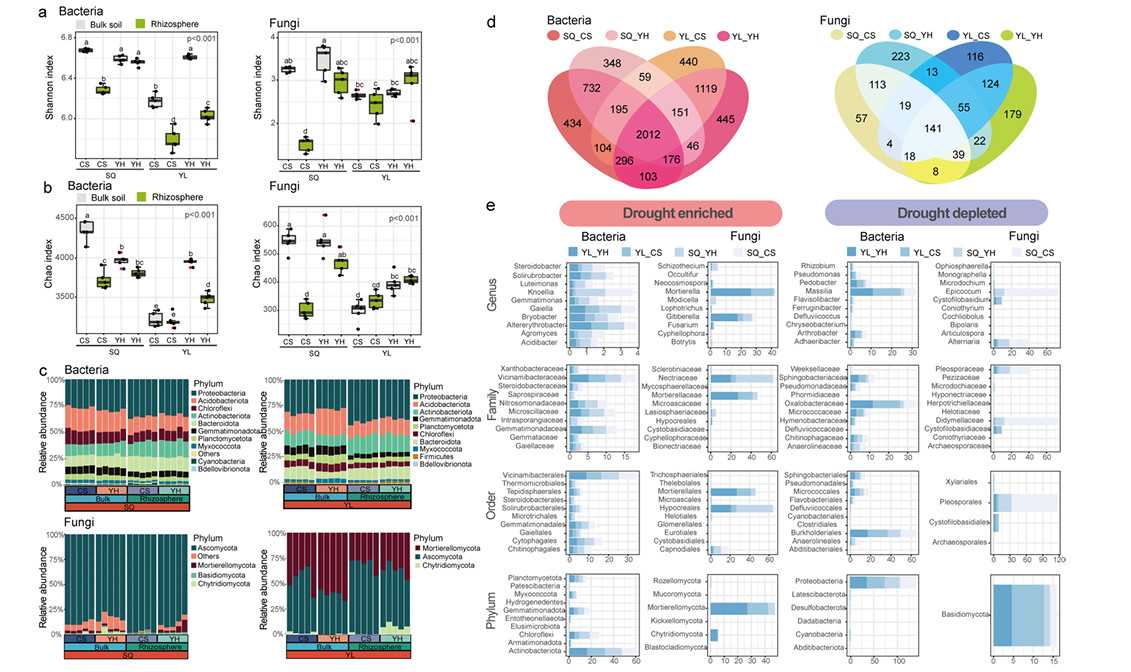 Patterns of diversity of soil microbiomes and influences of drought stress on enriched bacterial and fungal taxa (Yue et al., 2024)
Patterns of diversity of soil microbiomes and influences of drought stress on enriched bacterial and fungal taxa (Yue et al., 2024)
Discover the Microbial Function of Plant Freezing Stress Alleviation
Wild plants in alpine areas coexist with endophytic microbial communities, which may help plants grow and survive in cold conditions. In Rosaceae fruit crops, freezing stress is one of the main abiotic stresses affecting fruit yield, which often leads to the damage of flowers and leaves. Using or manipulating plant-related microbial communities is expected to be a sustainable method to alleviate the cold stress of crops, but the microbial mechanism of alleviating the cold stress of Rosaceae plants is still unclear. Amplicon sequencing, culture dependence and plant bioassay methods were integrated to reveal the structure and function of endophytic bacteria community in Rosaceae plants in the mountains, and the role of core groups in plant freezing stress tolerance.
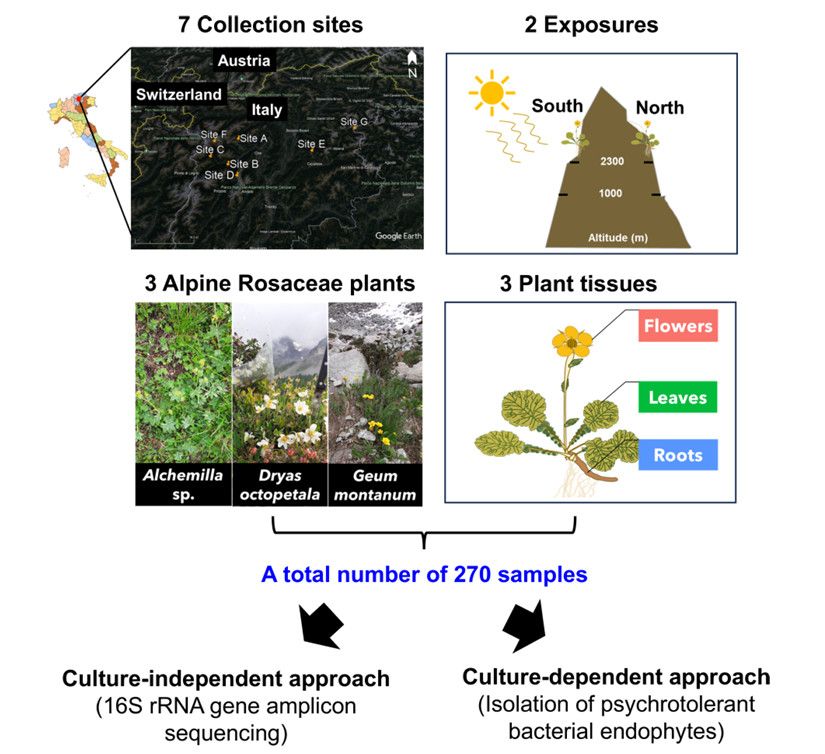 Experimental design of the research (Marian et al., 2025)
Experimental design of the research (Marian et al., 2025)
The 16S rRNA amplicon sequencing analysis of 270 samples showed that the endophytic bacterial community structure of Rosaceae plants in alpine region was significantly influenced by plant tissues, collection sites and host plants. The bacterial richness and α diversity of roots are higher than those of leaves and flowers, and there are significant differences in the community structure of endophytic bacteria in different plant tissues. Proteobacteria is a dominant phylum, and there are specific dominant families and genera in different tissues.
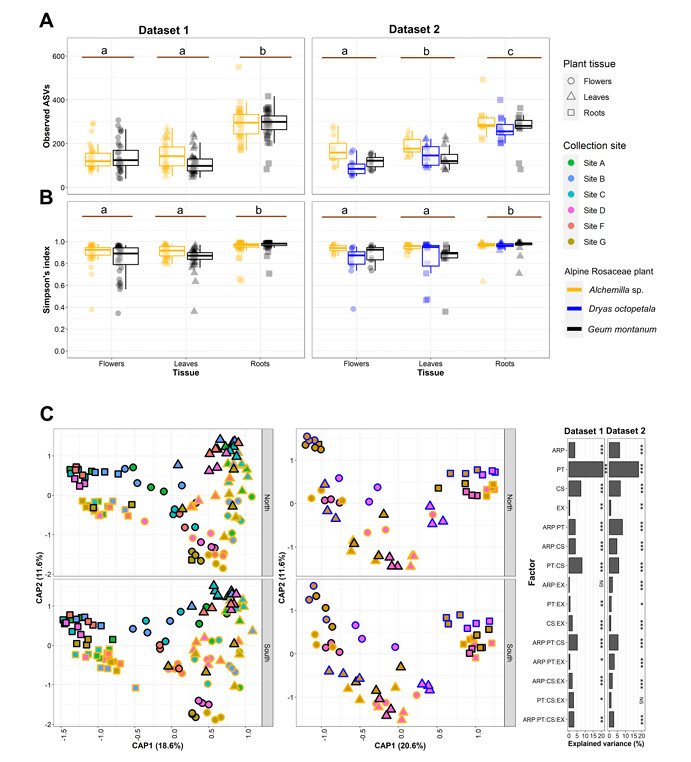 Diversity of endophytic bacterial communities associated with alpine Rosaceae plants (Marian et al., 2025)
Diversity of endophytic bacterial communities associated with alpine Rosaceae plants (Marian et al., 2025)
The core endophytic bacteria groups of Rosaceae plants in the alpine region were identified, including 31 highly common amplicon sequence variants (ASVs), accounting for 41% of the total relative abundance and 24% of the total β diversity based on Bray-Curtis differences. The core groups include cold-tolerant bacteria of Duganella, Erwinia, Pseudomonas and Rhizobium, which are ubiquitous in plant tissues, and some ASVs are predicted by Sloan neutral model of host selection.
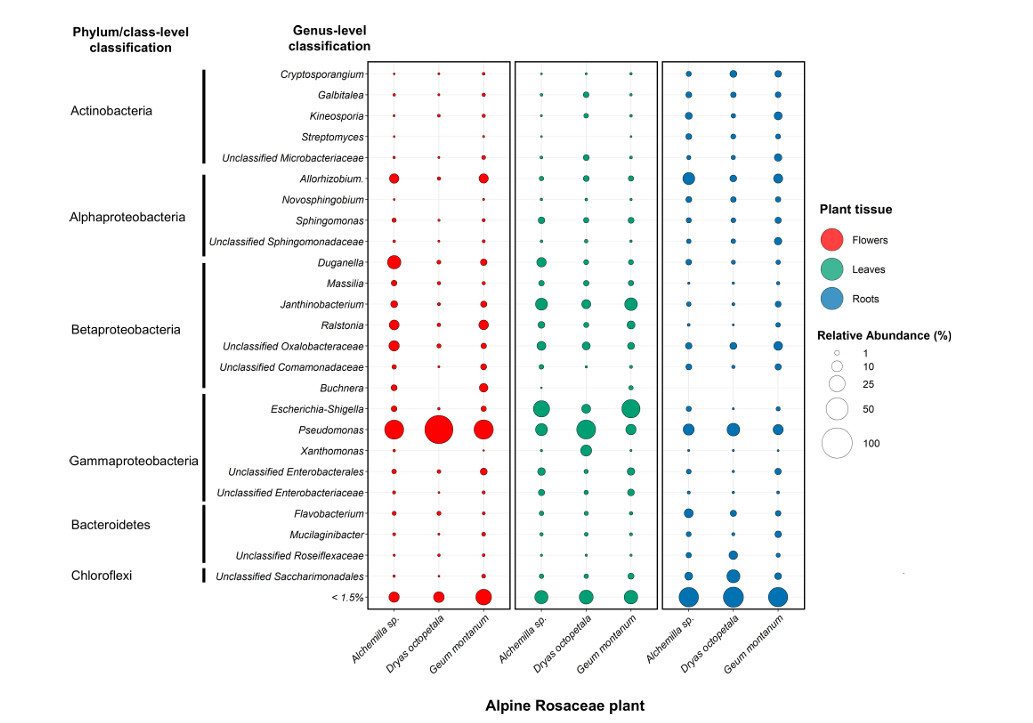 Taxonomic summary of the most abundant endophytic bacterial genera in alpine Rosaceae plants (Marian et al., 2025)
Taxonomic summary of the most abundant endophytic bacterial genera in alpine Rosaceae plants (Marian et al., 2025)
A large number of cold-tolerant culturable bacteria (685 strains) were isolated from Rosaceae plants in the mountains. These bacteria belong to four phyla and 65 genera. It was found that 91.5% of the representative bacteria could reduce the electrolyte leakage of strawberry seedlings under freezing stress, and the best strains (such as Duganella sp. GFBS205, Erwinia sp. GFBS303, etc.) belonged to the core group. These bacteria can colonize strawberry seedlings and survive at -6 ℃ in the treated plants.
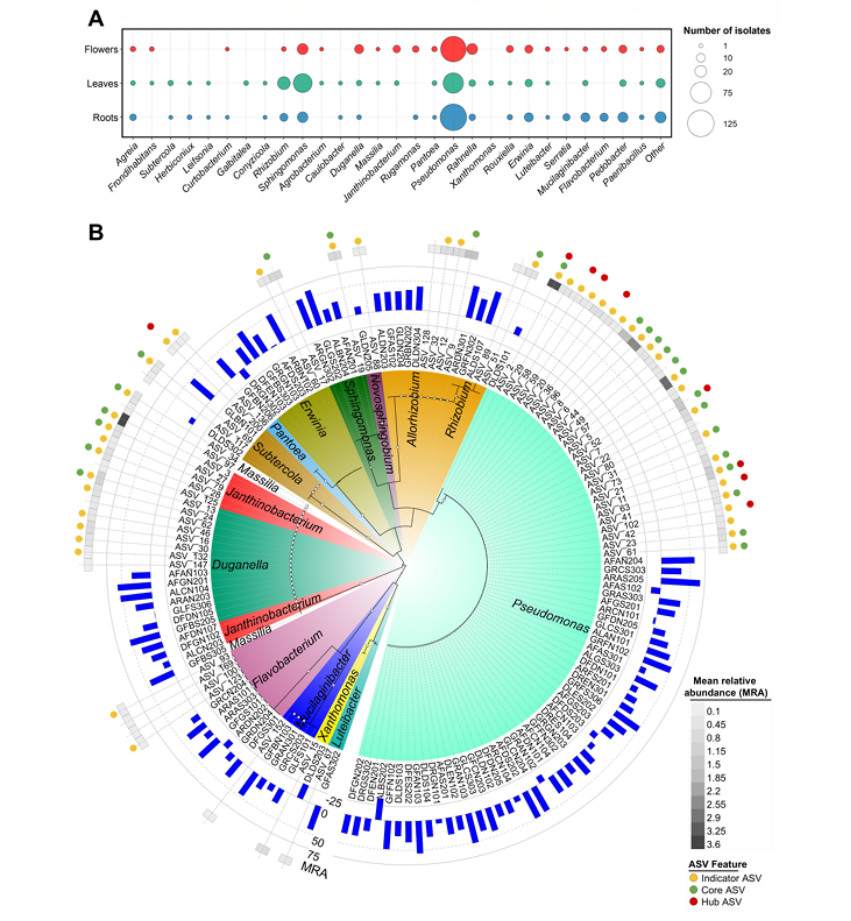 List of the psychrotolerant bacterial endophytes isolated from the selected alpine Rosaceae plants and their freezing protection ability (Marian et al., 2025)
List of the psychrotolerant bacterial endophytes isolated from the selected alpine Rosaceae plants and their freezing protection ability (Marian et al., 2025)
The research results provide theoretical basis and potential resources for developing biostimulants based on cold-tolerant endophytic bacteria to alleviate cold stress in agriculture, and are expected to provide new sustainable strategies for the management of frozen stress of Rosaceae fruit crops.
Identify Microorganisms Related to Plant Health
Ginger is a perennial monocotyledonous herb, and its underground rhizome has long been used as fresh vegetables, spices and medicinal materials. However, this crop is vulnerable to many plant pathogens, and root rot has become the main limiting factor of ginger production and market potential.
The purpose of this research is to deeply explore the microbial assembly related to plant health. By analyzing the soil and plant tissue samples of healthy and diseased ginger plants, the diversity of soil and endophytic microbial communities was evaluated, and the microorganisms related to plant health were further identified. By using non-targeted metabonomics technology, the metabolites related to plant health were identified, and the relationship between them and microbial communities was explored. Finally, it reveals how metabolites drive the assembly of healthy microbial communities, and then affect plant health.
The total number of bacterial genera in healthy plants is significantly higher (3331, of which 138 are unique genera), indicating that their microbial communities are richer. The number of bacterial genera in diseased plants decreased (2512, only 58 unique genera), but the number of bacterial genera in diseased plants was more except rhizosphere soil, which may be related to the enrichment of pathogenic bacteria or microorganisms adapted to the disease environment.
The total number of fungi in healthy plants (922 genera and 61 endemic genera) is higher than that in diseased plants (833 genera and 38 endemic genera), but the diversity of fungi in the rhizomes of diseased plants is higher (70 genera vs. 53 genera of healthy plants), which may be related to the colonization of pathogenic fungi.
The roots (588 genera) and rhizomes (550 genera) of healthy plants contain the most abundant endophytic bacteria, while the number of bacteria in these parts of diseased plants decreases sharply due to root rot (roots: 220 genera; Rhizome: 332 genera). The fungal diversity of diseased plant roots is higher, which may be related to the invasion or immunosuppression of pathogenic fungi (such as Gibellulopsis).
The bacterial diversity of healthy plants is dominant, and the fungal diversity of diseased plants is higher, which indicates that the stability of bacterial community may be related to disease resistance, and fungal proliferation may be a sign of disease aggravation.
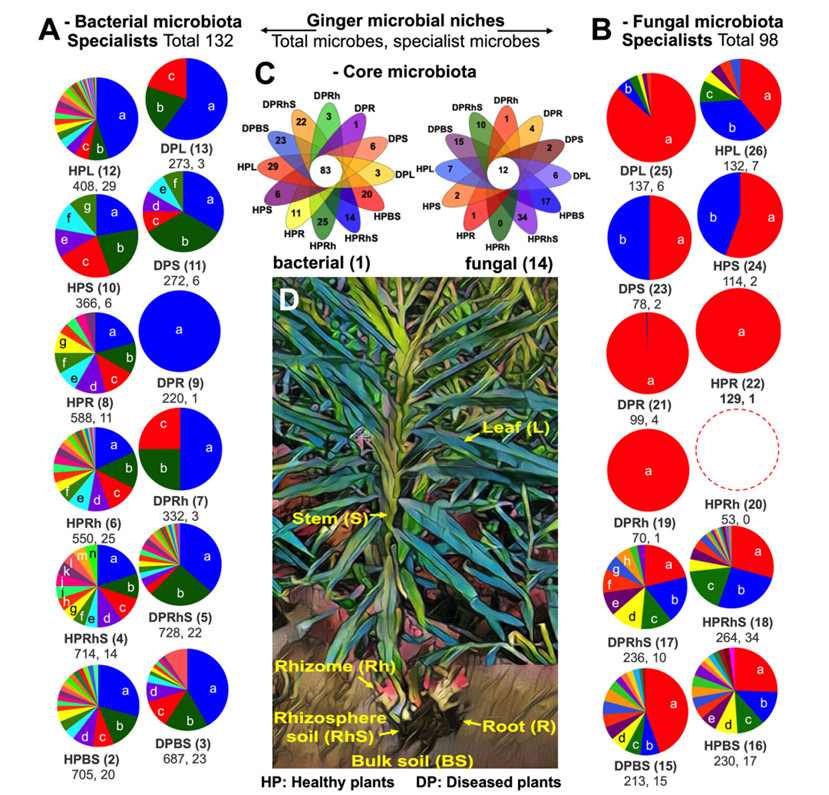 The plant disease rhizome rot drives changes in the assembly of the microbiota associated with the entire ginger plant (Wang et al., 2024)
The plant disease rhizome rot drives changes in the assembly of the microbiota associated with the entire ginger plant (Wang et al., 2024)
The root of ginger plant and ginger blast will significantly change the composition and diversity of microbial community, in which the diversity of bacteria decreases and the diversity of fungi increases. Compared with healthy plants, there are some bacterial and fungal species related to plant health in the soil and endophytic fungi community of diseased plants, which may be beneficial to plant health.
Challenges of Plant Long Amplicon Sequencing
Facing the complexity of plant genome and the limitation of technology itself, long amplicon sequencing still faces many problems to be solved in practical application.
Data Quality Problem
In the long amplicon sequencing of plants, the data quality is interfered by many factors. At the level of sequencing technology, PacBio and Nanopore platforms can produce very long reading sequences, but due to the lack of PCR amplification and error correction, single molecule sequencing is prone to random base mismatch, insertion or deletion errors.
In order to improve data quality, bioinformatics algorithm plays a key role. DeepConsensus algorithm based on deep learning can improve the accuracy of PacBio HiFi sequencing data from 99% to 99.99% by training a large number of sequencing data. In the aspect of experimental process optimization, touchdown PCR is used to adjust the annealing temperature, which can reduce primer dimer and nonspecific amplification. Increasing the sequencing depth to 30-50X, through the consistency analysis of multiple sequences, random errors can be greatly reduced.
Complexity of Data Analysis
The large amount of data and complex structure generated by long amplicon sequencing pose a challenge to traditional data analysis methods. The amount of data in a single sample is often hundreds of GB, and there are chimeras and high repetition regions in long read sequences, which makes the time-consuming of sequence alignment increase significantly. Taking the full-length sequencing of 16S rDNA as an example, when using BLAST to annotate species, the alignment time of long sequences is shorter and the reading sequence is extended by 3-5 times. In addition, function prediction needs to integrate multiple databases, but the existing databases are not suitable for long reading sequences, which leads to the lack of functional annotations of some genes.
Cost-effectiveness
The cost of long amplicon sequencing for plants mainly comes from sequencing reagents, equipment rental and data analysis resources consumption. In order to reduce the cost, we can start with experimental design and data analysis. In experimental design, stratified sampling strategy is adopted to reduce redundant samples. For example, in forest ecosystem research, spatial gradient sampling combined with random sampling can reduce the sample size by 40% and maintain data reliability.
Conclusion
Long Amplicon sequencing technology has become an important tool for plant science research because of its excellent ability in analyzing the structure and function of plant microbiome. From the perspective of development potential, this technology can help cultivate disease-resistant and stress-resistant crop varieties and optimize precise fertilization strategies in the agricultural field; In the field of ecology, it can provide a basis for dynamic monitoring of microbial communities for ecological restoration projects and promote the sustainable development of ecosystems.
Looking forward to the future, with the continuous innovation of sequencing technology and the continuous deepening of research, by optimizing the experimental process, developing efficient analytical tools and establishing industry standards, amplicon sequencing for long-term reading of plants will surely play a more critical role in the field of plant science and provide more powerful support for solving practical problems such as agricultural production and ecological protection.
References
- Yue H, Sun X, Wang T., et al. "Host genotype-specific rhizosphere fungus enhances drought resistance in wheat." Microbiome. 2024 12 (44) https://doi.org/10.1186/s40168-024-01770-8
- Marian M, Antonielli L, Pertot I, Perazzolli M. "Amplicon sequencing and culture-dependent approaches reveal core bacterial endophytes aiding freezing stress tolerance in alpine Rosaceae plants." mBio. 2025 16:e01418-24 https://doi.org/10.1128/mbio.01418-24
- Wang W, Portal-Gonzalez N, Wang X., et al. "Metabolome-driven microbiome assembly determining the health of ginger crop (Zingiber officinale L. Roscoe) against rhizome rot." Microbiome. 2024 12 (167) https://doi.org/10.1186/s40168-024-01885-y
For research purposes only, not intended for personal diagnosis, clinical testing, or health assessment