
 Sample Submission Guidelines
Sample Submission Guidelines
The result of the Human Genome Project is a recognized "standard" for scientists – the reference genome. Like a coordinate system, researchers can better understand the structure, function, and variation of the human genome against the reference genome. The Human Genome Project began in 1990, the working draft was published in 2001, and the final sequencing map was completed in 2003, a landmark achievement in the history of human life sciences. However, this "map" is not complete, leaving 8% of the gaps.
The Missing 8% of the Human Genome Sequence
This 8% gap is mainly a heterochromatin region in the genome. The term heterochromatin is relative to euchromatin. Euchromatin is stained lightly by basic dyes or weakly positive for Forgan reaction; heterochromatin is stained darkly or positive for Forgan reaction. Heterochromatin is either condensed throughout the cell cycle, e.g., at sites such as mitoses, telomeres, and nucleolus-forming regions, or condensed from euchromatin to heterochromatin under certain developmental and physiological conditions, e.g., one of a pair of X chromosomes in female mammals condenses into relatively inactivated bacilli at day 16-18 of embryonic development.
The high degree of heterochromatin coalescence, along with being full of repetitive sequences, made sequencing difficult and did not allow for the assembly of sequencing results. This was due to the fact that the technology at that time only enabled short read sequencing, i.e., sequencing required breaking the genome into shorter DNA fragments, and then gradually splicing the fragments into the complete genome sequence by comparing and matching overlapping sequences through certain algorithms after completing sequencing of each fragment. Therefore, the existence of repetitive sequences greatly hinders the splicing and assembly of the fragments, and short read-length sequencing means that the sequenced DNA fragments must be short enough, just like using very small pieces for a puzzle, which further increases the difficulty of reconstructing the genome.
However, as human genome sequencing technology advances, the "pieces" are becoming larger and the "puzzle" less difficult to solve. Telomere (T2T) consortium announced a complete human genome sequence, T2T-CHM13, which fills the 8% gap. The genome sequence is derived from a Complete Hydatidiform Mole (CHM), which contains only the paternal genome and not the maternal genome. This allows the reconstruction of the genome without facing the difficulty of mis-assembling the paternal and maternal genomes by crossover, but also makes the final result fail to contain the Y chromosome sequence.
For more information about the T2T human genome, you can refer to the article The Complete T2T Genome by Sequencing Opens the Door to Post-Genomic Era.
A New Human Pan-Genome Draft
On May 10, the first human pangenome reference sketch produced by the Human Pangenome Reference Consortium (HPRC) was published in Nature. Unlike previous reference genomes, this pan-genome sketch contains a total of 94 genomes from 47 individuals with different ancestors from around the world, and the parents of these 47 individuals were also collected to analyze the paternal and maternal origins of their genes.
These 47 individuals included individuals from Africa, America and Asia, with representatives from China as well. Many of these samples are from the 1000 Genomes Project and are being re-analyzed by the Human Pan-genome Consortium using new technologies of "long-read sequencing". The team plans to expand the sample size to 350 individuals by mid-2024.
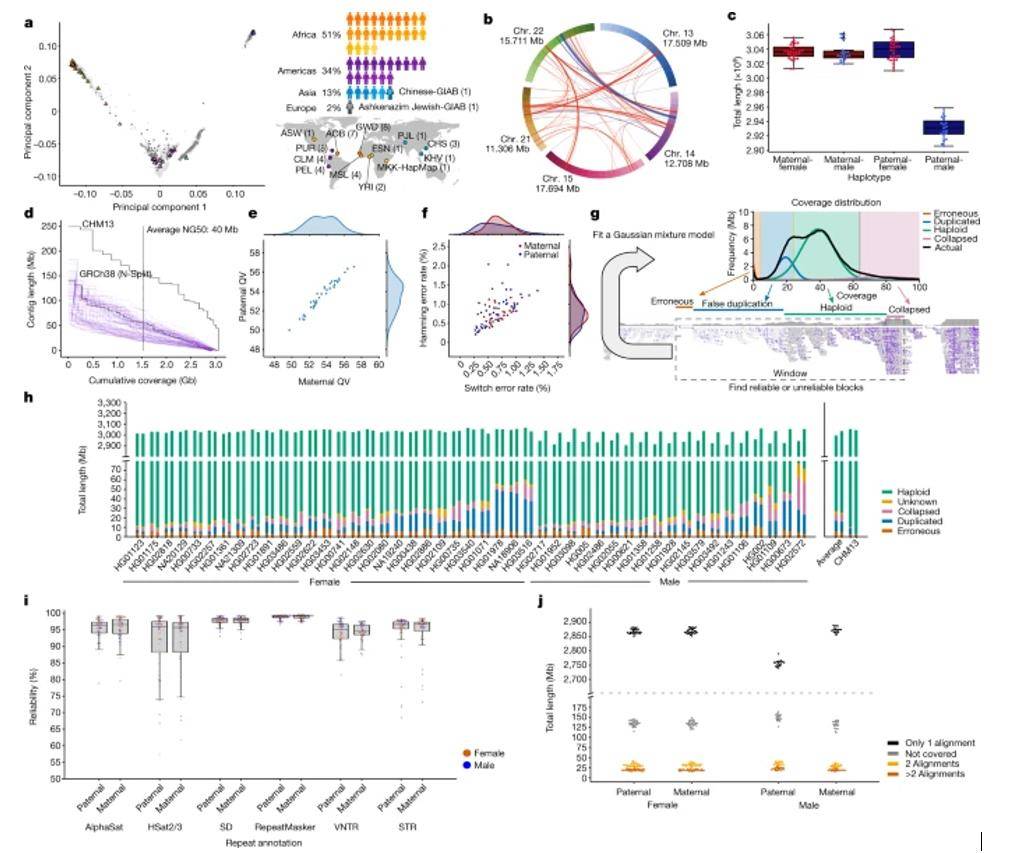
Selecting the HPRC samples. (Liao et al., 2023)
Compared to GRCh38, the results added 119 million base pairs to the original 3.2 billion base pairs, of which approximately 90 million are from structural variants. In addition, the team identified 1115 new gene repeats associated with evolution.
To better present the pan-genomic reference sketch, geneticist Eimear Kenny and her colleagues at the Icahn School of Medicine at Mount Sinai in New York computationally aligned the sequences of 47 individuals to create a "pan-genomic map" somewhat similar to a subway route map.
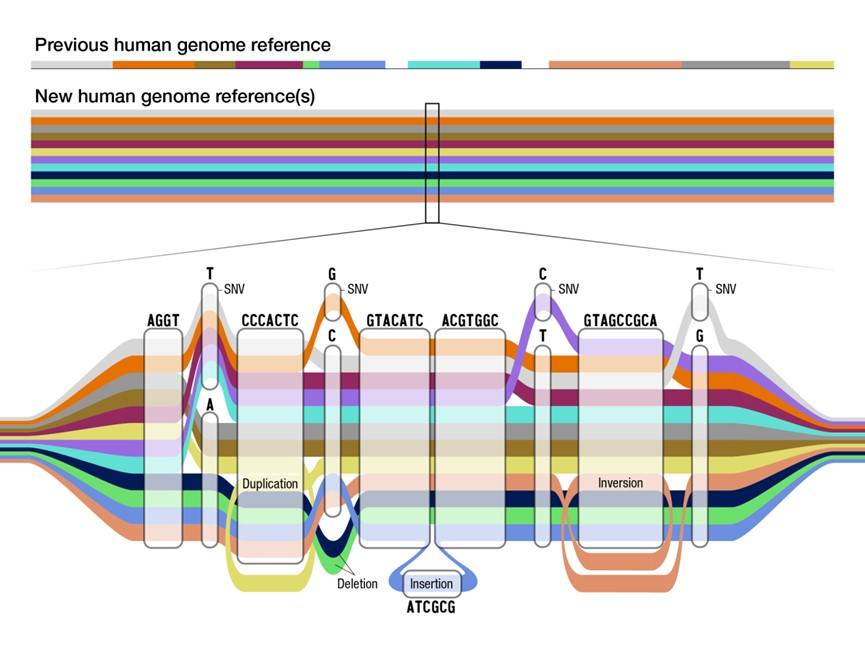
Human pan-genome reference.
On this map, the base identical parts remain linear as in the previous single genome reference, while divergences in routes appear where certain individual nucleotides differ, in addition to bypassing a nucleotide to represent a deletion (as in the green and dark blue paths above), wrapping clockwise to represent a duplication (as in the yellow path), wrapping counterclockwise to represent an inversion (as in the pink path), etc.
This time the pan-genomic reference sketch achieved 99% coverage of the expected sequence and also achieved over 99% accuracy at the structure and base pair level. Compared to the GRCh38-based workflow, using this sketch to analyze short read-length data reduces small genetic variation discovery errors by 34% and increases detection rates by 104% when detecting haplotype structural variants.
The human pan-genomic reference has allowed us to characterize tens of thousands of novel genomic variants that were previously unavailable," said Wen-Wei Liao, first author of the paper and a doctoral student at Yale University’s research affiliate and Washington University in St. Louis. With the pan-genomic reference, we can improve our understanding of the link between genes and disease, thereby accelerating clinical research."
Eric Green, M.D., director of the National Human Genome Research Institute (NHGRI), said, "Basic researchers, or clinicians, should have access to reference sequences that reflect the diversity of the population in order to reduce inequities in human health. The creation and updating of the human pan-genome is consistent with NHGRI’s consistent goal of pursuing global diversity in all aspects of genomics research, helping to advance the dissemination of genomic knowledge and promoting the implementation of genomic medicine in a more equitable manner."
Currently, most participating HPRC institutions are located in the U.S. and Europe, and HPRC member institution Karen Miga, a geneticist at the University of California, Santa Cruz, said the next phase of the project will move toward a truly international collaboration with adequate sampling and sequencing of historically underrepresented regions. In addition to using diverse genomes, the project will actively reach out to sample populations and understand their health care needs, allowing these populations to benefit directly from the project’s results.
References
- Liao, Wen-Wei, et al. "A draft human pangenome reference." Nature 617.7960 (2023): 312-324.
- Nurk S, Koren S, Rhie A, et al. The complete sequence of a human genome. Science, 2022, 376(6588): 44-53.