
 Sample Submission Guidelines
Sample Submission Guidelines
The Discovery of Hepatitis C Virus
Hepatitis, or the inflammation of the liver, is primarily caused by viral infection. Other significant factors to it include alcohol abuse, environmental contaminants, and autoimmune diseases. It became apparent in the 1940s that there were two major forms of contagious hepatitis. In the 1960s, Hepatitis B and A vaccines were being made but according to Harvey J. Alter, large cases of transfusion-related hepatitis still remained. This led to the identification of an unknown infectious agent that caused the “non-A, non- B” Hepatitis which is the untreated transfusion-related hepatitis cases. For more than a decade, the unknown infections agent was then identified. The phenomenal discovery that resulted in the 2020 Nobel Prize in Physiology or Medicine was awarded to the three scientists: Harvey J. Alter, Michael Houghton, and Charles M. Rice. Their landmark studies revealed that it was the Hepatitis C that was the culprit in the remaining blood-borne hepatitis. Harvey J. Alter’s analytical research of transfusion-associated hepatitis confirmed that a common cause of chronic hepatitis was an unknown virus. In order to separate the genome of a new virus called the Hepatitis C virus, Michael Houghton used an unestablished method. Charles M. Rice offered the final proof that the hepatitis C virus itself could develop hepatitis. The discovery of the Hepatitis C virus identified the cause of the remaining cases of chronic blood transfusion-related hepatitis and made it possible for thousands of lives to be saved by blood testing and new treatments.
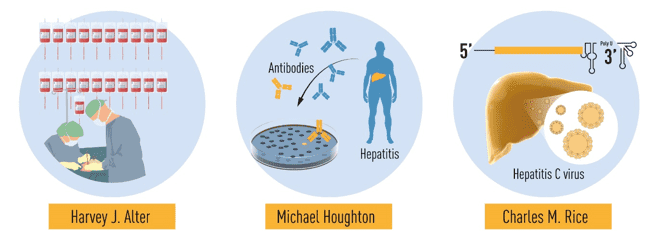
Figure 1. Summary of the discoveries awarded by this year’s Nobel prize (Karlen, 2020)
Detecting HCV through sequencing
The hepatitis C virus (HCV) has infected over 170 million people worldwide and is a globally relevant blood-borne agent that causes liver diseases. HCV is a diverse group of RNA viruses that are currently split into both genotypes 1-7 and subtypes. HCV infection can be treated with antiviral drugs, but it is important to establish the HCV genotype for optimal treatment strategy selection.
- PacBio sequencing
- Nanopore sequencing
Recently, newly developed novel sequencing technologies have been capable of producing exceptionally long reads. Single-Molecule Real-Time (SMRT) sequencing using the PacBio RSII or Sequel sequencers (Pacific Biosciences) is among the main third-generation sequencing technologies. SMRT sequencing is a system for sequencing-by-synthesis focused on real-time visualization of fluorescently tagged nucleotides along with the production of specific DNA model molecules. SMRT sequencing has been applicable to numerous scientific issues, such as identifying the full-length genome sequences of bacteria, metagenomic analysis of viruses or bacteria, haplotype gene determination, transcriptomic analysis of splicing variations, and determination of the full-length human genome sequence, taking the benefit of ultra-long read length into account.
Nanopore sequencing is another technique for single-molecule real-time sequencing. Nanopore sequencing does not use base synthesis reaction, which radically varies from other sequencing technologies. Instead, it specifically detects the sequence of the nucleotides that make up single-stranded DNA molecules by changing the electrical voltage as it moves through a protein pore.
Techniques for virus sequencing
Virus genome sequencing has been a valuable instrument for further determining the infectivity of viruses and immunological surveillance. Due to the low amount of virus genetic material relative to the host DNA, and the complexity of having a correct genome assembly, it is still a difficult task to extract the virus genome sequence immediately from clinical samples.
In metagenomic sequencing, complete DNA (and/or RNA) is derived and sequenced from a sample like a host, but also bacteria, viruses, and fungi. This method is quick and low-cost, and it is the only method that does not include comparison sequences. The disadvantage of metagenomic sequencing is that to acquire sufficiently viral genome content, it needs a very high sequencing depth.
The target enrichment sequencing utilizes oligonucleotides for virus-specific capture to enhance the processing of the viral genome before sequencing. This approach is more precise than metagenomics sequencing but means greater prices for sample processing and more specialized scientific knowledge.
A well-established approach consisting of precise viral genome amplification by PCR before sequencing is PCR amplicon sequencing. In regular use, it is conveniently useful in a variety of samples and is thus very suitable for clinical samples. For samples with very low viral genetic content, the PCR amplification approach is especially important.
References:
- Mauriera F., Beurya D., Fléchonb L., et al. A complete protocol for whole-genome sequencing of virus from clinical samples: Application to coronavirus OC43. 2019.
- Ogata N., Alter H. J., Miller R. H, et al. Nucleotide sequence and mutation rate of the H strain of hepatitis C virus. Proceedings of the National Academy of Sciences, 1991, 88(8), 3392–3396.
- Takeda H, Yamashita T, Ueda Y, et al. Exploring the hepatitis C virus genome using single molecule real-time sequencing. World J Gastroenterol. 2019, 25(32): 4661-4672.
- Virtanen E., Mannonen L., Lappalainen M., et al. Genotyping of hepatitis C virus by nucleotide sequencing: A robust method for a diagnostic laboratory. MethodsX, 2018, 5, 414–418.