
 Sample Submission Guidelines
Sample Submission Guidelines
The use of PD-1/PD-L1 inhibitors has significantly improved overall survival in a subset of non-small cell lung cancer (NSCLC) patients, but only about 20% of patients respond to treatment. Identifying predictors of response is crucial to improve patient selection for immunotherapy. However, current understanding of relevant predictors is limited by the lack of large, multi-omic studies specific for immunotherapy in NSCLC.
A recent study published in Nature Genetics by the Massachusetts General Hospital Cancer Center, Broad Institute and AstraZeneca team addressed this gap by performing a joint analysis of the Stand Up To Cancer-Mark Foundation (SU2C-MARK) cohort. The study identified multiple molecular signatures associated with patient response outcomes, including favorable (ATM mutations) and unfavorable (TERT amplification) genomic signatures. Additionally, the study found an association between expression of immunoproteasome inducible components and response, and explored the impact of intrinsic tumor subtypes.
Cohort mutation profile
The study highlights the complexity of the biological determinants behind immunotherapy response in NSCLC, and the potential for comprehensive analysis in large, specific cancer cohorts to improve patient selection for treatment.
The study analyzed tumor samples from 393 patients with advanced non-small cell lung cancer (NSCLC) before they received first-line treatment with PD-1/PD-L1 drugs. Both tumor and matched normal samples underwent whole exome sequencing (WES), and some tumor tissue also underwent RNA-seq analysis. The researchers assessed the prevalence of known lung cancer drivers in three remission categories (PR/CR, SD, and PD) and found that non-synonymous TMB was associated with the remission category. They also analyzed the relationship between 49 known lung cancer drivers and treatment response, identifying six genes that were statistically significant or near-significant, including ATM mutations, which appeared to be most favorable for checkpoint inhibition therapy.
In addition, the team performed transcriptional predictor identification of response and genome-wide analysis of differentially expressed genes between responders (PR/CR) and non-responders (SD/PD), initially identifying three associated genes: PSME1, PSME2 and PSMB9. Further exploration of genes specific to the antigen presentation pathway showed that immunoproteasome subunits are important predictors of therapeutic response, even in the broader context of IFN-γ-induced transcription. The results suggest that individual driver events may define favorable and unfavorable factors for immune checkpoint inhibitor therapy beyond TMB metrics.
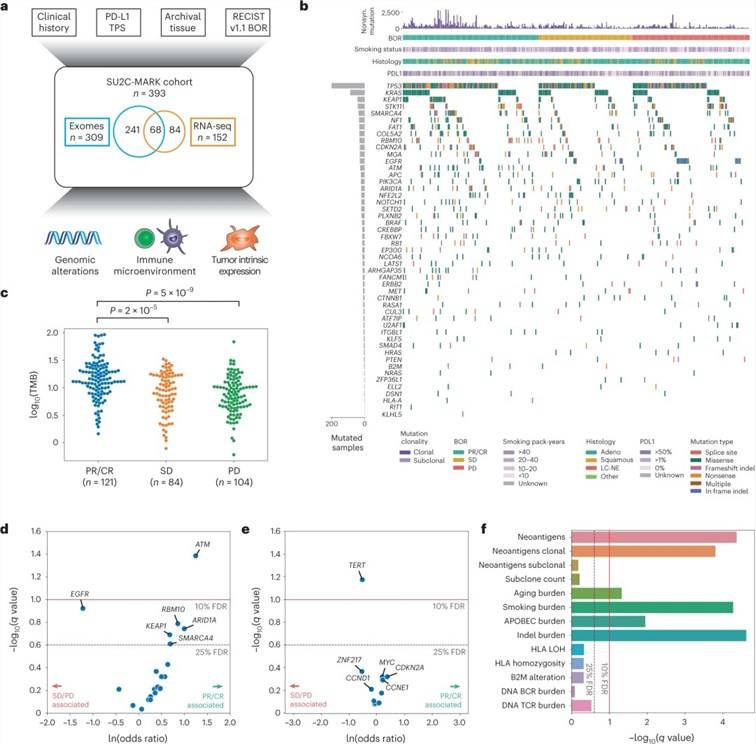
Overview of clinical and genomic data collected across the SU2C-MARK cohort. ()
Microenvironmental expression signatures
Researchers used Bayesian non-negative matrix decomposition (B-NMF) to identify microenvironmental (M) signatures associated with immunotherapeutic responses. The researchers identified three distinct M signatures (M-1, M-2, and M-3) based on the differential expression of 770 genes, which were derived from sequencing data of mixed cell populations.
Gene set enrichment analysis was then used to determine the functional associations of each M signature. M-1 was found to be associated with epithelial-mesenchymal transition, while M-2 was associated with allograft rejection/IFN-γ response, indicating an inflammatory immune environment. M-3 was weakly associated with cell cycle-related E2F targets.
The researchers found that these subtypes had different response rates to immune checkpoint inhibitors, with M-2 showing a significantly increased response rate relative to M-1 and M-3. Overall, this study suggests that M signatures can be useful in predicting responses to immunotherapy and may provide insights into the underlying biology of the tumor microenvironment.
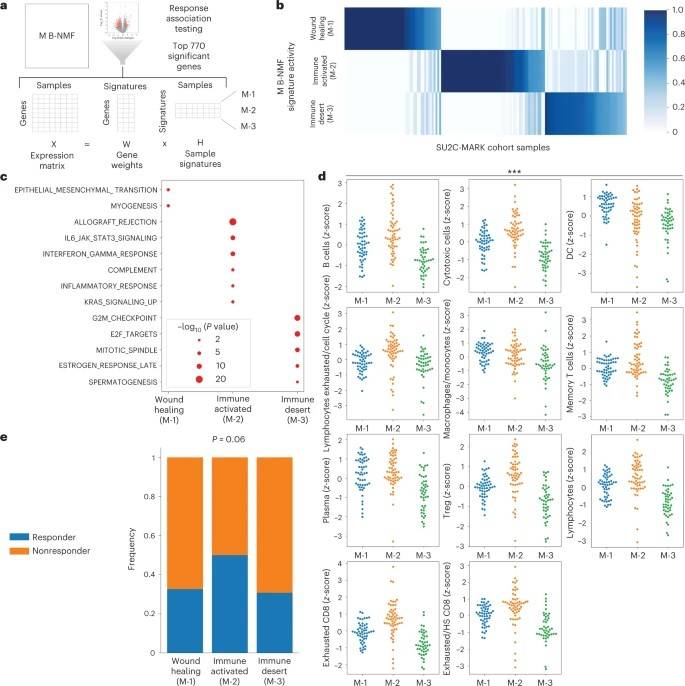
Overview of M signature generation using B-NMF. ()
Comprehensive analysis of the study cohort
The researchers performed a comprehensive analysis of the relationships between predictors of checkpoint blockade responses in NSCLC. They obtained three strong correlation modules and a fourth group enriched for single-gene alterations.
The first correlation module (C1) appears to reflect the "wound healing" microenvironment, which includes immunosuppressive myeloid cells and stromal features. The second correlation module (C2) reflects the more classical cytokine and immune environment associated with "immune activation/failure," including infiltrative immune features and proteasome subunits. The third correlation module (C3) consists of features associated with mutational burden and may be representative of neoantigen abundance and subsequent enhanced immune recognition.
The fourth group (C4) is enriched for single-gene alterations with different immunobiological features. Notably, this module includes EGFR mutations, which show little correlation with immune features but moderate correlation with mutation load features. This suggests that intrinsic resistance in this subtype may be primarily driven by neoantigen deficiency.
Overall, the comprehensive analysis highlights the complex interplay between various clinical, genomic, and transcriptomic features in checkpoint blockade responses in NSCLC. The identification of these correlation modules provides insights into potential mechanisms underlying response and resistance to checkpoint blockade therapy and may inform the development of more effective personalized treatment strategies for NSCLC patients.
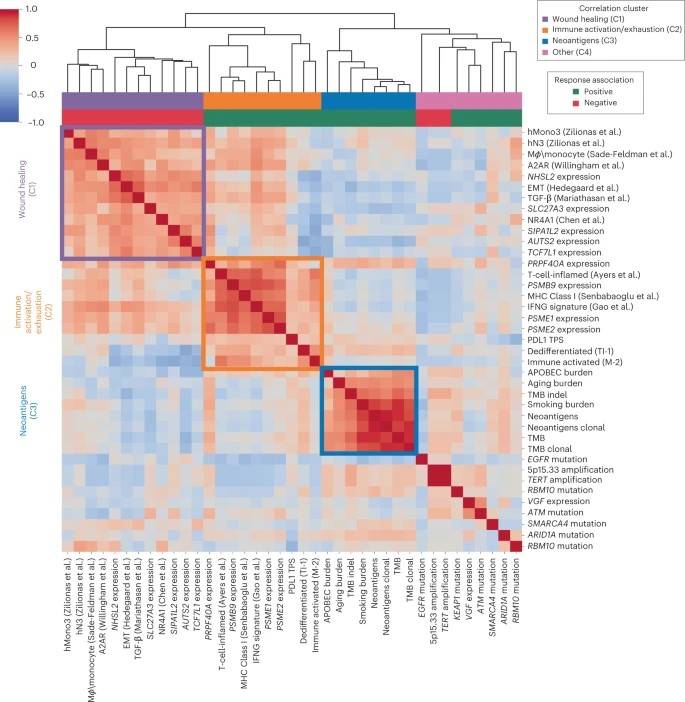
Cross-correlation heatmap of the top response and resistance-associated features. ()
In Summary
The study found that the expression of EMT (epithelial-to-mesenchymal transition) and TGF-b (transforming growth factor beta) signaling was mainly observed in fibroblasts and endothelial cells within the tumor microenvironment, rather than in the mesenchymal epigenetic status of the tumor cells themselves. The study also identified specific single-gene transcriptional predictors, such as AUTS2 and TCF7L1, which were substantially expressed in the tumor cells themselves.
Overall, the study suggests that the response and resistance to immune checkpoint inhibitors may be driven by a complex ecosystem of interactions between different cellular components within the tumor microenvironment. The findings highlight the importance of understanding the specific signaling pathways and cell types involved in response and resistance to immune checkpoint inhibitors, which may help in the development of more effective interventions for NSCLC.
This study analyzed the SU2C-MARK cohort of non-small cell lung cancer (NSCLC) patients to identify different predictors associated with immunotherapy response. The analysis included genomic features, immune-related features, and tumor biology-related features, revealing complex interactions between different signaling pathways and cell types. The researchers hope that the cohort will continue to be a resource for further research on the genomic predictors of immunotherapy response and antitumor immunobiology.
Reference
- Ravi A, Hellmann M D, Arniella M B, et al. Genomic and transcriptomic analysis of checkpoint blockade response in advanced non-small cell lung cancer[J]. Nature Genetics, 2023: 1-13.