Unveiling Transcriptome Complexity with Iso-Seq Analysis
At a glance:
- 1. Introduction
- 2. Iso-Seq Workflow
- 3. Advantages of Iso-Seq
- 4. Applications of Iso-Seq
- 5. Computational Innovations and Challenges
- 6. Future Directions
- 7. Conclusion
Using full-length RNA sequencing, Iso-Seq technology overcomes short-read assembly challenges, enabling detailed profiling of transcriptomic complexity. This method accurately maps alternative splicing, gene fusion events, and novel isoforms, deepening our understanding of gene regulation and cellular function. Iso-Seq's versatility spans oncology, immunology, neurobiology, and agricultural genomics, where it provides critical insights into disease processes and adaptive responses. Combined with advanced bioinformatics and cutting-edge AI methodologies, Iso-Seq further precision medicine by facilitating biomarker discovery and targeted therapy development. Its innovative approach enhances transcript annotation and promises significant advances in personalized healthcare and genomic research. Overall, Iso-Seq revolutionizes modern transcriptomic analysis today.
Introduction
Complexity of the Transcriptome for Isoform Profiling
The transcriptome constitutes the complete repertoire of RNA species present within a cell or organism, encompassing messenger RNAs (mRNAs), various non-coding RNAs (ncRNAs), and regulatory RNAs. This dynamic network is continuously modulated by extrinsic stimuli, endogenous signals, and developmental transitions. Fundamental molecular mechanisms-such as alternative splicing, alternative polyadenylation, gene fusions, and diverse post-transcriptional modifications-generate an extensive array of transcript isoforms. Each isoform contributes uniquely to cellular function and adds successive layers of complexity to gene expression regulation.
A profound comprehension of this multifaceted RNA landscape is indispensable for elucidating the molecular mechanisms governing cellular behavior and disease progression. The heterogeneity produced by alternative splicing is central to myriad biological functions and is inextricably linked to pathologies including oncogenesis, neurodegeneration, and genetic disorders. Consequently, precise isoform profiling is essential for decoding gene expression dynamics and identifying potential targets for therapeutic intervention.
Limitations of Conventional Short-Read RNA Sequencing
While RNA sequencing (RNA-Seq) has revolutionized transcriptomic studies, traditional short-read RNA-Seq techniques are inherently limited in resolving the full spectrum of isoform diversity. Typically, these methods generate reads shorter than 300 nucleotides, necessitating elaborate computational assembly to reconstruct complete transcripts. Such assembly processes frequently falter when confronted with genes that exhibit intricate splicing architectures-especially those with numerous exons, overlapping transcriptional units, or nested splice junctions. For example, the DSCAM gene in Drosophila, which can potentially generate thousands of isoforms, epitomizes the challenges inherent to assembling short-read data.
Moreover, short-read methodologies struggle to differentiate isoforms in genomic regions characterized by high repetitiveness or segmental duplications. Rare or transient transcripts may be underrepresented due to limited sequencing depth, and structural variants, such as fusion transcripts commonly observed in neoplastic cells, are prone to misassembly or complete omission. Collectively, these limitations impede the accurate capture of transcriptomic diversity, thereby constraining our understanding of gene regulation and hindering the identification of robust biomarkers.
Iso-Seq: A Long-Read Sequencing Solution
To overcome these challenges, Isoform Sequencing (Iso-Seq) has emerged as a revolutionary methodology that harnesses long-read sequencing technology. Developed by Pacific Biosciences, Iso-Seq employs Single-Molecule Real-Time(SMRT) sequencing to directly read full-length RNA molecules-from the 5' cap to the poly-A tail-thus obviating the need for fragment assembly. The contiguous reads produced by Iso-Seq offer an accurate and comprehensive representation of transcript isoforms.
Iso-Seq confers several significant advantages. It enables the precise delineation of alternative splicing events, including subtle variations such as alternative donor/acceptor site usage and intron retention. In addition, Iso-Seq facilitates the detection of novel isoforms emerging from previously unannotated exons, antisense transcription, or readthrough events. Importantly, this technology effectively resolves complex genomic regions-such as those with tandem repeats or overlapping genes-thereby substantially enhancing reference annotations. As such, Iso-Seq has established itself as a cornerstone in functional genomics, revealing previously concealed layers of transcriptional complexity.
Iso-Seq Workflow
Library Construction
The Iso-Seq workflow initiates with the synthesis of full-length complementary DNA (cDNA) from high-quality, intact RNA. Reverse transcription is conducted using template-switching oligonucleotides to ensure complete conversion of RNA molecules into cDNA, thereby preserving the native 5' and 3' ends. This initial step is critical for capturing the entirety of each transcript and maintaining the inherent complexity of the transcriptome.
Subsequent PCR amplification is meticulously optimized to minimize bias while preserving the relative abundance of rare isoforms. The amplified cDNA is then enzymatically processed into circularized templates, known as SMRTbell libraries, which are specifically engineered for compatibility with PacBio's sequencing chemistry. These circular constructs facilitate continuous, real-time sequencing of both strands, enhancing consensus accuracy and ensuring a faithful representation of full-length transcripts.
SMRT Sequencing
At the core of the Iso-Seq methodology lies SMRT sequencing, which operates within nanostructures known as Zero-Mode Waveguides (ZMWs). In these confined environments, individual DNA polymerases are immobilized, and as they synthesize complementary strands, fluorescently labeled nucleotides are incorporated and detected in real time. This direct observation yields long, uninterrupted reads, typically ranging from 10 to 20 kilobases, with some reads surpassing 50 kilobases under optimized conditions.
A key advantage of SMRT sequencing is its avoidance of PCR amplification biases, thereby preserving the true stoichiometry of the original RNA population. The inherent single-molecule resolution of this technology provides an unbiased and precise snapshot of the transcriptome, which is indispensable for accurately identifying complex isoforms.
Data Processing
Following sequencing, raw subreads generated from individual ZMWs undergo Circular Consensus Sequencing (CCS). This iterative process re-sequences the same molecule multiple times to correct stochastic errors, yielding high-fidelity (HiFi) reads with accuracies exceeding 99.9%. These HiFi reads offer a near-exact representation of the original RNA molecules.
Subsequently, HiFi reads are clustered into isoform families using specialized algorithms, such as those implemented in IsoSeq3. This clustering step consolidates redundant sequences while preserving distinct splice variants, thereby facilitating the precise identification of alternative splicing events, novel isoforms, and transcript variants. The refined resolution achieved through this approach is critical for downstream analyses and comprehensive transcriptome annotation.
Bioinformatics Analysis
The final phase of the Iso-Seq workflow involves rigorous bioinformatics analysis. Isoform sequences are aligned to reference genomes using splice-aware tools such as GMAP, which accommodate both canonical and non-canonical splice junctions as well as structural variations. Concurrently, SQANTI3 classifies isoforms into categories (e.g., full-splice matches, novel isoforms) and filters out potential artifacts.
Furthermore, integration of Iso-Seq data with conventional short-read RNA-Seq datasets-via pipelines employing tools like TAMA and TAPIS-facilitates accurate quantification of isoform expression across samples. This integrative strategy not only refines genome annotations, particularly in species with limited reference data, but also provides a holistic view of transcriptomic diversity, thereby advancing our understanding of context-specific splicing regulation.
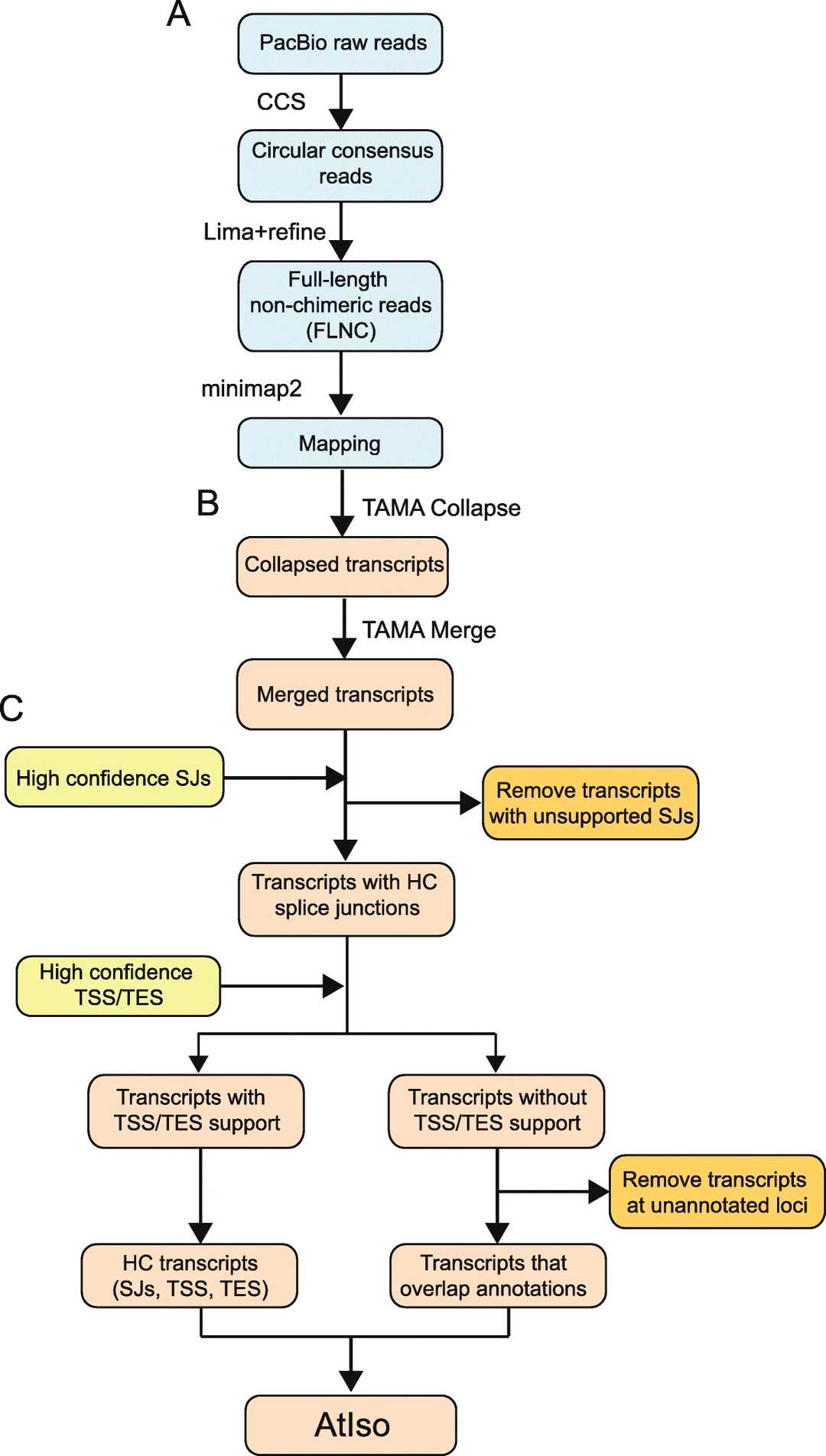 Figur1 . Iso-Seq analysis workflow.(Zhang, R. et al, 2022)
Figur1 . Iso-Seq analysis workflow.(Zhang, R. et al, 2022)
Advantages of Iso-Seq
Unparalleled Resolution of Transcript Structures
Iso-Seq's ability to sequence entire transcripts in a single, continuous read obviates the need for computational assembly, thereby eliminating the ambiguities inherent in short-read approaches. This direct sequencing method ensures that even the most complex isoforms are fully captured, resulting in a more accurate and comprehensive depiction of the transcriptome. For instance, investigations of human brain tissue have revealed thousands of previously uncharacterized isoforms, thereby significantly enriching the current understanding of neural gene regulation.
Deciphering Complex Splicing Patterns in Disease
The high resolution provided by Iso-Seq enables the detection of intricate splicing events, including exon skipping, intron retention, and alternative splice site usage. These splicing events are critical determinants of gene function and have profound implications for disease pathology. In oncological research, Iso-Seq has uncovered cryptic splicing events that contribute to tumorigenesis and therapeutic resistance, highlighting its essential role in elucidating the molecular basis of disease.
Discovery of Novel Isoforms and Fusion Transcripts
The long-read capabilities of Iso-Seq render it particularly effective for the identification of novel isoforms and fusion transcripts-variants frequently associated with pathological conditions such as cancer. These previously uncharacterized transcript variants, often overlooked by short-read sequencing, present new opportunities for biomarker discovery and the development of targeted therapies. By providing a comprehensive view of transcript diversity, Iso-Seq substantially broadens the scope of functional genomics research.
Precision in Allele-Specific and Epitranscriptomic Analyses
The high-fidelity reads generated by Iso-Seq facilitate the precise detection of single-nucleotide polymorphisms (SNPs) and enable robust analyses of allele-specific expression. Such precision is indispensable for understanding genetic variation and its implications for disease susceptibility and personalized treatment strategies. Additionally, Iso-Seq can capture RNA modifications through kinetic analysis, thereby enriching the emerging field of epitranscriptomics and offering further insights into gene regulation.
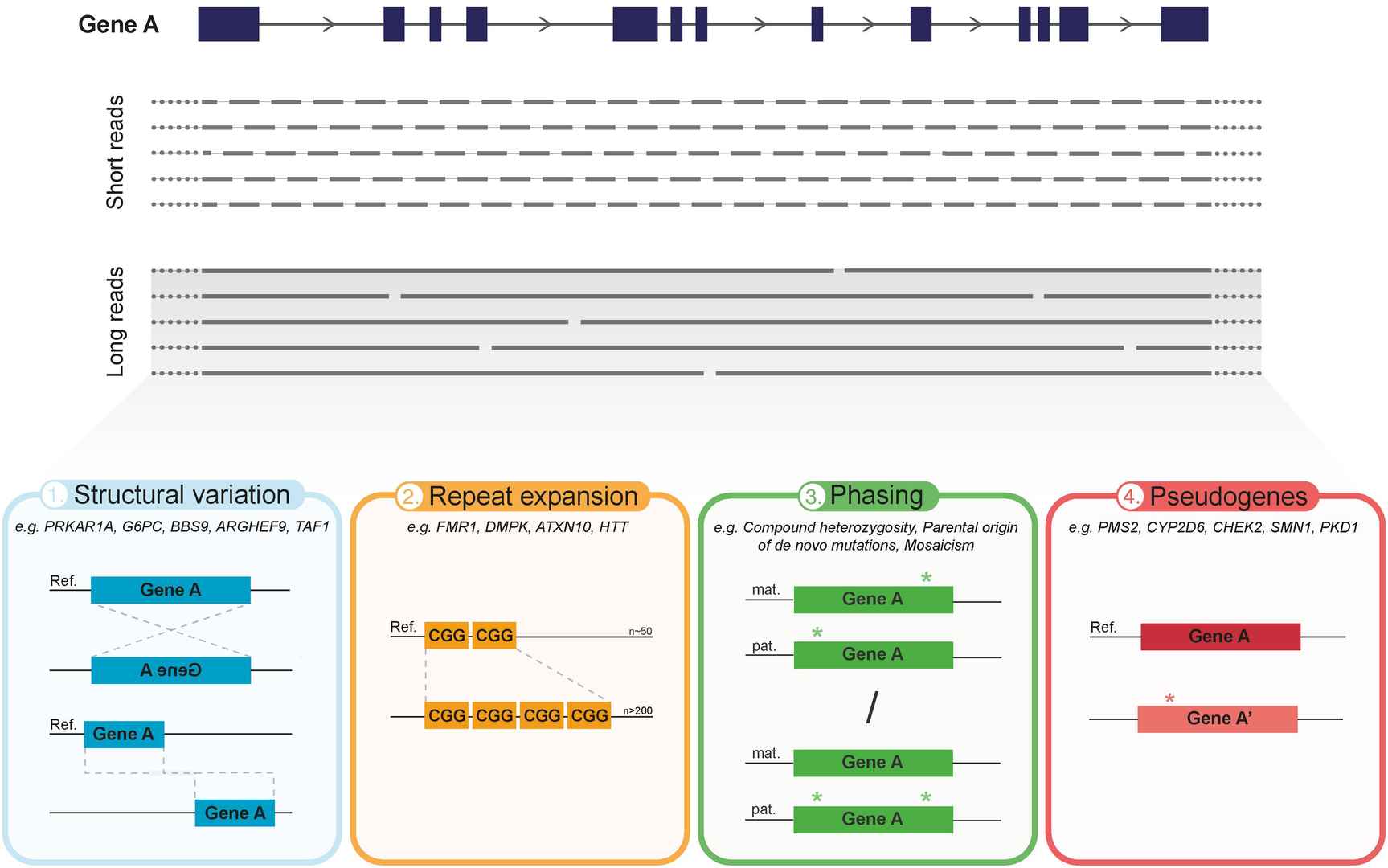 Figur2 . Advantages of Iso-Seq analysis.(Mantere, S. et al, 2019)
Figur2 . Advantages of Iso-Seq analysis.(Mantere, S. et al, 2019)
Applications of Iso-Seq
Oncology: From Isoform Discovery to Targeted Therapeutics
In oncology, Iso-Seq has redefined the transcriptomic landscape by revealing oncogenic isoforms and fusion transcripts that drive tumor progression. For example, the identification of the EML4-ALK fusion in lung adenocarcinoma has provided critical insights into tumor biology and informed the development of targeted therapeutic interventions. Furthermore, the detection of neoantigens-aberrant peptides generated through alternative splicing-has opened new avenues for the development of personalized cancer vaccines, underscoring the profound clinical implications of Iso-Seq.
Immunology: Elucidating Adaptive Immune Repertoires
Iso-Seq has proven invaluable in immunological research by enabling the comprehensive profiling of full-length T-cell and B-cell receptor transcripts. This capability allows for the detailed assessment of clonal diversity and somatic hypermutation, which are essential for understanding the adaptive immune response. Such insights have significant implications for the design and development of advanced immunotherapies, particularly in the treatment of autoimmune diseases and cancer.
Neurobiology: Mapping Isoform Diversity in the Brain
The human brain is characterized by remarkable transcriptomic complexity, with genes such as DSCAM and NRXN1 exhibiting extensive alternative splicing that is critical for neural connectivity and function. Iso-Seq has been instrumental in mapping this diversity, revealing splice variants that correlate with neurological conditions such as schizophrenia and epilepsy. Detailed isoform maps generated by Iso-Seq contribute to a deeper understanding of neural circuitry and may facilitate the discovery of novel targets for therapeutic intervention in neurodegenerative disorders.
Agricultural and Ecological Genomics
Iso-Seq also finds extensive application in agricultural and ecological genomics. In crop research, Iso-Seq has been employed to resolve the complex transcriptomes of species such as maize, enabling the identification of stress-responsive isoforms that are vital for drought tolerance and yield improvement. Similarly, in ecological studies, Iso-Seq has elucidated adaptive splicing mechanisms in organisms subjected to environmental extremes, such as coral species under thermal stress. These applications illustrate the broad utility of Iso-Seq across diverse biological fields.
Computational Innovations and Challenges
SMRT Link: An Integrated Data Processing Ecosystem
Central to the Iso-Seq methodology is the SMRT Link software suite, which provides a comprehensive environment for raw data processing, base calling, and error correction. Recent enhancements, such as GPU acceleration for Circular Consensus Sequencing (CCS), have markedly reduced processing times while ensuring the generation of high-fidelity reads. This integrated ecosystem is pivotal for maintaining the accuracy and reliability of Iso-Seq datasets.
Machine Learning for Advanced Isoform Annotation
Emerging computational tools incorporating machine learning, such as IsoAnnot, are beginning to play a crucial role in the annotation of isoforms. These algorithms are capable of discriminating between transcriptional noise and isoforms with distinct functional roles, as observed in key genes like TP53, where truncated variants may exhibit divergent biological activities. Such innovations promise to refine isoform annotation further, thereby deepening our understanding of transcript functionality.
Overcoming Cost and Throughput Constraints
Historically, the implementation of Iso-Seq necessitated significant resources, often requiring multiple SMRT cells per sample. However, the advent of the Revio platform has dramatically increased sequencing throughput while substantially reducing costs. This technological advancement is democratizing access to long-read sequencing, enabling a broader range of research institutions and clinical laboratories to adopt Iso-Seq for diverse applications.
Future Directions
Enhanced Throughput with the Revio Platform
The forthcoming Revio sequencing system is anticipated to further enhance Iso-Seq by significantly increasing throughput and reducing per-sample expenses. This development will enable large-scale transcriptomic studies, thereby extending the application of Iso-Seq across both basic research and clinical contexts, particularly in fields such as oncology, immunology, and neurobiology.
Integration with Single-Cell and Spatial Transcriptomics
Future research endeavors are expected to integrate Iso-Seq with single-cell RNA sequencing (scRNA-Seq) and spatial transcriptomics. By combining the comprehensive, full-length transcript information provided by Iso-Seq with the cellular resolution of scRNA-Seq and the spatial context offered by spatial transcriptomics, researchers will gain unprecedented insights into cell-type-specific gene regulation, tissue organization, and disease progression. This integrative approach is poised to revolutionize our understanding of developmental processes and pathophysiological mechanisms.
AI-Driven Enhancements in Isoform Prediction
Advancements in artificial intelligence and machine learning are set to further refine the interpretation of Iso-Seq data. Future algorithms will likely improve isoform prediction accuracy, streamline the annotation of complex splicing events, and facilitate rapid identification of novel transcript variants. These enhancements will accelerate transcript discovery and expand our understanding of gene function and regulatory networks.
Expanding Clinical and Translational Applications
Iso-Seq is positioned to become a cornerstone of precision medicine. By accurately identifying disease-specific isoforms and splicing biomarkers, Iso-Seq offers substantial potential for informing targeted therapeutic strategies, monitoring disease progression, and personalizing treatment regimens. Its capability to detect subtle variations in gene expression and RNA modifications may lead to more effective clinical interventions for a broad spectrum of diseases, ranging from cancer to neurodegenerative disorders.
Conclusion
Iso-Seq represents a transformative advance in transcriptomic research, providing unparalleled resolution in characterizing isoform diversity and gene regulation. By directly sequencing full-length transcripts and accurately capturing complex splicing events, Iso-Seq overcomes many limitations inherent in conventional short-read RNA sequencing. This breakthrough technology has significantly enriched the understanding of fundamental biological processes and holds immense promise for clinical applications, from biomarker discovery to the development of targeted therapeutics.
As sequencing technologies and bioinformatics tools continue to evolve, the capabilities of Iso-Seq are expected to expand further. Future integrations with single-cell and spatial transcriptomics, coupled with advancements in machine learning, are poised to revolutionize the study of gene expression at the cellular level. In both basic research and clinical practice, Iso-Seq is rapidly establishing itself as an indispensable tool for elucidating the complexities of the transcriptome.
In summary, the advancements enabled by Iso-Seq constitute a substantial leap forward in our ability to profile and interpret the full landscape of RNA molecules. As this technology becomes increasingly accessible and its applications continue to diversify, it is set to redefine the standards of transcriptomic research and contribute significantly to the progress of precision medicine.
References
- T. Mantere, S. Kersten, A. Hoischen, Long-Read Sequencing Emerging in Medical Genetics. Front Genet 10, 426 (2019). https://doi.org/10.3389/fgene.2019.00426
- R. Zhang et al., A high-resolution single-molecule sequencing-based Arabidopsis transcriptome using novel methods of Iso-seq analysis. Genome Biol 23, 149 (2022). https://doi.org/10.1186/s13059-022-02711-0
For research purposes only, not intended for personal diagnosis, clinical testing, or health assessment