
AAV ITR Sequencing: Unraveling the Workflow, Result Analysis, Technical Hurdles, and Future Trends
Adeno-associated virus (AAV), as a star vector of gene therapy, has shown great potential in the treatment of central nervous system diseases, retinopathy, muscular dystrophy and various organ dysfunction due to its excellent safety, low immunogenicity, wide host range and stable gene expression characteristics.
The reverse terminal repeat (ITR) of AAV, as the core regulatory element of key links such as virus replication, packaging, genome integration and escape, has become the key to unlock the full therapeutic potential of AAV. However, due to its high GC content and complex secondary structure, ITR brings great challenges to sequencing. This text expounds its sequencing process, results analysis methods, technical challenges, and future development trends.
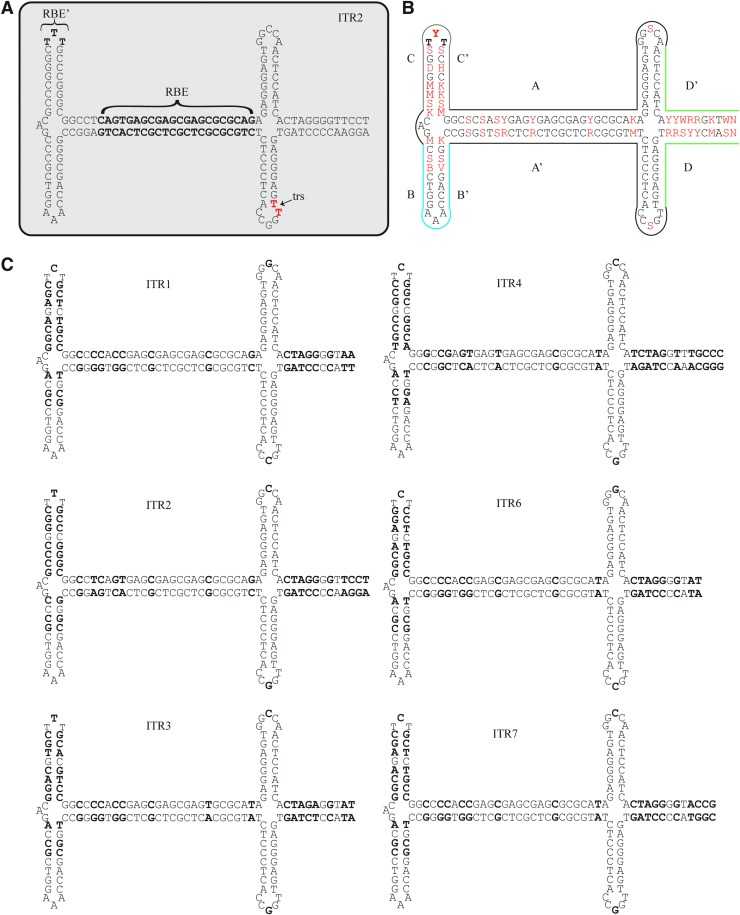 Sequence and structures of AAV ITRs from serotypes 1-4, 6, and 7 (Earley et al., 2020)
Sequence and structures of AAV ITRs from serotypes 1-4, 6, and 7 (Earley et al., 2020)
AAV ITR Sequencing Workflow
ITR of AAV is very important for the replication, packaging and integration of virus genome. Accurate determination of ITR sequence is helpful to understand the biological characteristics of AAV and is of great significance in the construction of gene therapy vectors. The workflow of AAV ITR sequencing is described in detail below.
Sample Preparation
To obtain samples containing AAV from the production system, ultracentrifugation and column chromatography can be used to separate AAV virus particles from other impurities (such as host cell proteins, nucleic acids, etc.) to obtain high-purity AAV samples. Using nucleic acid extraction kit or related reagents, the purified AAV virus particles are cracked to release viral genomic DNA, and pollutants such as protein and polysaccharide are removed to obtain pure AAV genomic DNA as a template for subsequent PCR amplification.
PCR Reaction
According to the known conserved sequence of AAV ITR, specific PCR primers were designed. Primers should be highly specific and only amplify the ITR region to avoid non-specific amplification of other viruses or host genome fragments.
A proper amount of AAV genomic DNA template, upstream and downstream primers, deoxyribonucleoside triphosphate (dNTPs), DNA polymerase and PCR buffer were added to the PCR reaction tube in turn. The concentration of each component needs to be optimized to ensure the efficiency and specificity of PCR reaction.
After the PCR reaction, a small amount of products were analyzed by agarose gel electrophoresis. Electrophoresis at a suitable voltage for a period of time, observed by gel imaging system, if a clear and single band appears in the expected position, it indicates that the ITR region has been successfully amplified.
Construction of Sequencing Library
Using PCR product purification kit, impurities such as primer dimer, unreacted dNTPs and redundant primers in PCR reaction system were removed to obtain pure ITR PCR products.
The purified PCR products were repaired by related enzymes to make the ends of double-stranded DNA flush. Subsequently, a prominent A base was added to the 3' end to connect with the sequencing linker with a T-protruding end.
The sequencing linker with specific sequence and function was connected with the PCR product with A tail. The linker not only provides primer binding sites for subsequent sequencing reactions, but also carries index sequences for sample identification. The ligation products were enriched by PCR amplification to further increase the number of target ITR sequences in the library.
High Throughput Sequencing
The constructed sequencing library is loaded on the sequencer for sequencing according to the standard flow of the selected sequencing platform. During the sequencing process, the instrument will monitor the sequencing reaction in real time and record the base incorporation in each cycle, thus generating a large number of sequencing data. During the sequencing process, experimental conditions, such as temperature and humidity, should be strictly controlled to ensure the stability and accuracy of sequencing results.
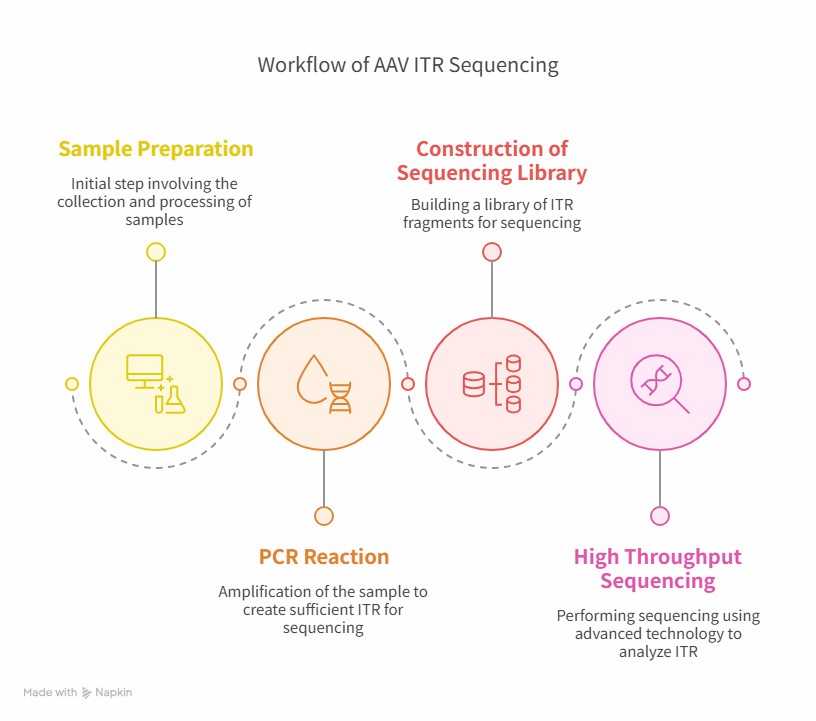 Workflow of ITR sequencing
Workflow of ITR sequencing
Take the Next Step: Explore Related Services
Learn More
- AAV in Gene Therapy: Rationale, Mechanisms, Applications, Safety, and Efficiency Enhancement
- In-Depth Exploration of AAV: From Biological Traits to Gene Therapy Horizons
- Key Aspects of AAV Capsid Protein: Structure, Function, Engineering and Applications
- Viral Therapy Explained: Concepts, Classifications, and Clinical Applications
How to Analyze ITR Sequencing Results
It is a key task to deeply analyze the sequencing results of ITR. This process plays an indispensable role in understanding viral genome structure, transposon behavior and many phenomena related to gene function and regulation.
Gene Sequence Pattern Recognition ITR
ITR has a unique sequence pattern, which usually shows a reverse complementary repetitive structure. With the help of special bioinformatics algorithms and tools, ITR regions can be accurately identified in the compared sequences according to these characteristic patterns. These tools determine the boundary and specific sequence content of ITR by strictly searching and matching the reverse complementarity of sequences. According to the typical structural characteristics of ITR, researchers can further verify the accuracy of the identification results and ensure that key ITR areas will not be misjudged or missed.
Analysis of ITR Length and Sequence Variation
After determining the ITR region, the length and sequence variation were deeply analyzed. The ITR length of different species or individuals of the same species may be different, which is often closely related to gene function, virus infection characteristics or transposon activity. Through the statistical analysis of ITR length of multiple samples, the length polymorphism at the population level can be revealed. At the same time, using sequence analysis software, the base differences between ITR sequences of different samples were carefully compared, and the variation types such as single nucleotide polymorphism (SNP) and insertion deletion (InDel) were identified. These variation information is of great significance for understanding the evolution mechanism of ITR in the evolution process and its influence on related biological processes.
Correlation Analysis with Functional Components
ITR is not isolated in the genome, and it often has close interaction with peripheral functional elements, such as promoters, enhancers or coding genes. Through the integrated analysis of genome annotation information, researchers can determine the relative position relationship between ITR and these functional elements. If ITR is close to the promoter region of a gene, it may imply that it plays an important role in the transcription regulation of the gene. Further combining with gene expression data, such as RNA sequencing results, studying the relationship between regional variation of ITR and changes in gene expression level can deeply reveal the regulation mechanism of ITR on gene function and provide key clues for comprehensive understanding of biological processes.
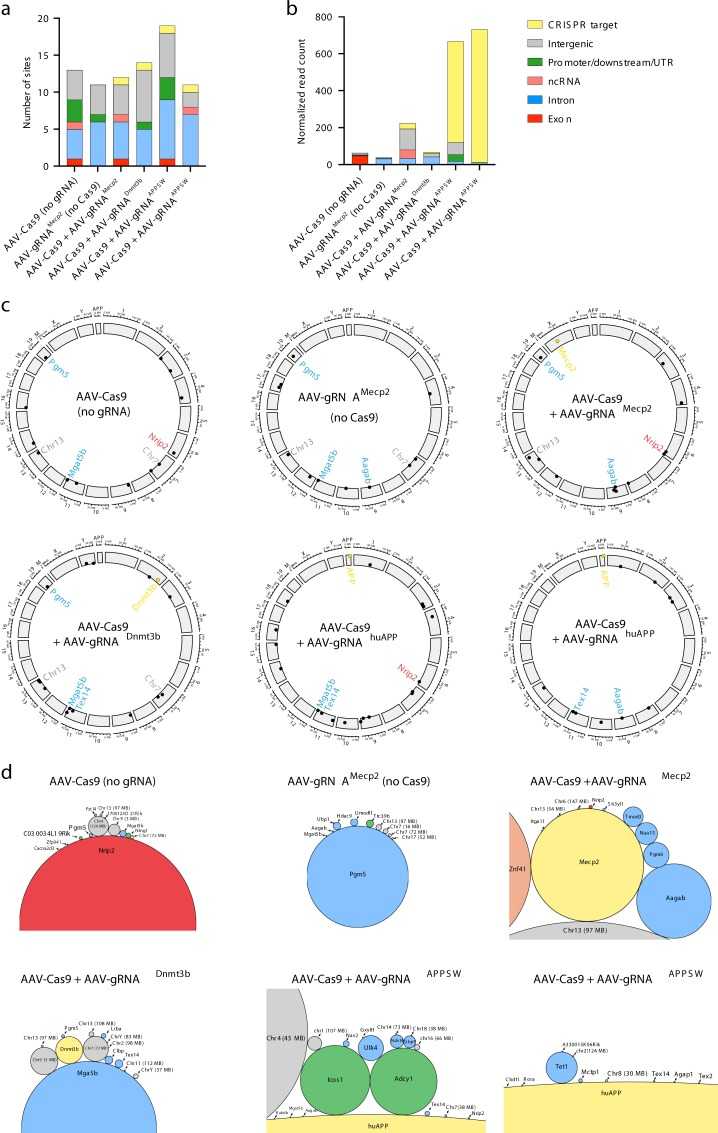 Genome-wide AAV mapping from CRISPR treated mouse brains (Hanlon et al., 2019)
Genome-wide AAV mapping from CRISPR treated mouse brains (Hanlon et al., 2019)
AAV ITR Sequencing Challenges
ITR not only plays a central role in the replication and packaging process of AAV, but also has a far-reaching impact on the stability, transduction efficiency and safety of the carrier. However, due to its special structure and sequence characteristics, the accurate sequencing of ITR faces many technical challenges.
High Palindrome Structure
AAV ITR has a unique high palindrome structure, which makes DNA double-stranded easy to form hairpin structure under certain conditions. When DNA polymerase encounters hairpin structure during sequencing, it may stop, slide or extend incorrectly. Because the hairpin structure will hinder the smooth movement of polymerase along the template chain, the sequencing signal will be interrupted or inaccurate base addition will occur, so that the sequencing results will appear peak trapping, signal weakening or even unreadable.
Repetitive Sequence
There are repeated sequences in ITR, which further increases the complexity of sequencing. Short tandem repeats are prone to mismatch and amplification deviation during PCR amplification. Due to the similarity of repeat units, PCR primers may bind nonspecific to different repeat sites, resulting in the amplification of a variety of products with different lengths. In the second generation sequencing (NGS), these amplification products with different lengths will be sequenced at the same time, which makes it difficult to accurately distinguish the real ITR sequence from the artifacts caused by amplification deviation in data analysis, and reduces the quality and reliability of sequencing data.
Purity of AAV Carrier
In practical application, AAV vector may contain impurities, such as host cell DNA, residual plasmid DNA or protein. These impurities will interfere with the sequencing reaction. For example, it may be difficult to completely separate host cell DNA from AAV DNA in the extraction process, and a large number of background signals of host cell DNA will mask the weak signal of AAV ITR during sequencing, resulting in inaccurate sequencing results. If the residual plasmid DNA contains sequences similar to ITR, it may compete with AAV ITR in the process of PCR amplification or library construction, which will affect the effective amplification and sequencing of AAV ITR.
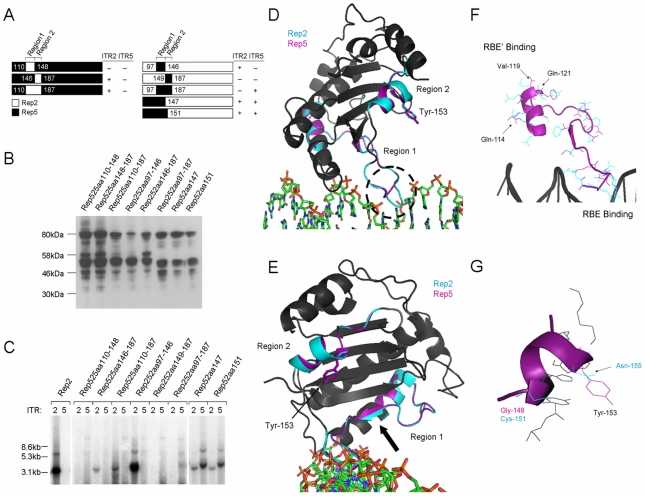 Characterization of Rep regions critical for ITR specificity (Hewitt et al., 2010)
Characterization of Rep regions critical for ITR specificity (Hewitt et al., 2010)
Future Development of AAV ITR Sequencing
Developing advanced sequencing technology that can accurately detect ITR sequence and deeply analyze the relationship between its structure and function is of great significance for promoting the wide application of AAV vector in gene therapy and ensuring the quality and safety of gene therapy products.
Optimized Data Analysis
In order to solve the problems of short reading length and difficult splicing of the second generation sequencing technology in ITR sequencing, researchers are actively developing a sequence splicing algorithm specifically for the complex structure of ITR. These algorithms fully consider the secondary structure characteristics of ITR region, high GC content distribution and known sequence conservative patterns, and adopt more intelligent and efficient splicing strategies, which significantly improve the accuracy and integrity of ITR region sequence splicing.
In addition to the sequence splicing algorithm, a series of bioinformatics tools for identifying the regional variation and structural characteristics of ITR are also emerging. These tools can deeply mine and analyze a large number of data obtained by sequencing, and accurately identify information such as point mutation, insertion/deletion mutation, structural variation and potential functional elements in ITR region.
ITR Comprehensive and Accurate Sequencing
In order to give full play to the advantages of different sequencing technologies and achieve comprehensive and accurate sequencing of AAV ITR region, it is a promising strategy to combine long-reading and short-reading sequencing technologies. Specifically, the complete frame sequence of ITR region can be obtained by using three generations of long reading and long sequencing technology (such as SMRT sequencing or Nanopore sequencing) to determine its overall structure and approximate variation; Then, the second generation short reading length sequencing technology is used to perform high-depth sequencing on the ITR region, and the low-frequency variation and tiny mutation that may be missed by long reading length sequencing can be accurately detected by virtue of its Qualcomm quantity. By integrating, analyzing, complementing and verifying the data obtained by the two technologies, the sequence information of ITR region can be analyzed more comprehensively and accurately.
Developing New Sequencing Technology
With the continuous progress of science and technology, a new generation of sequencing technology with better performance is expected to appear in the future to meet the complex needs of AAV ITR sequencing. These new technologies may further improve the accuracy and throughput of sequencing while maintaining the advantages of long reading length, while reducing the error rate and noise signal in the sequencing process.
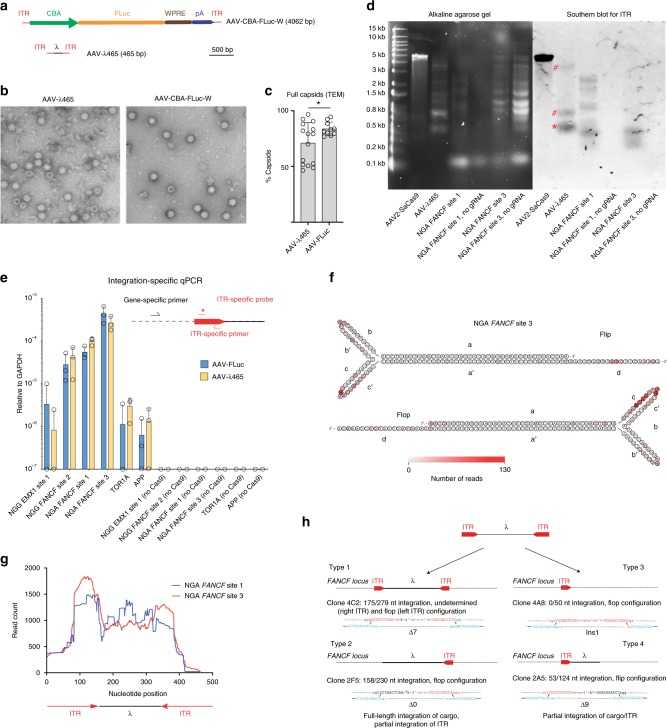 Characterization of AAV vector integration into CRISPR cut sites using a miniaturized AAV genome (Hanlon et al., 2019)
Characterization of AAV vector integration into CRISPR cut sites using a miniaturized AAV genome (Hanlon et al., 2019)
Conclusion
To sum up, the ITR of AAV plays a central role in the life cycle and gene therapy application of AAV. The unique structural features of ITR, including palindrome sequence, high GC content and T-shaped hairpin structure, endow it with important functions, but also lead to its instability and easy truncation and mutation during plasmid amplification, which poses a severe challenge to the production and quality control of AAV vectors.
Although a lot of progress has been made in AAV-ITR sequencing and vector optimization, there is still a broad space for exploration. In the future, it is necessary to further study the interaction mechanism between ITR and virus proteins and host cytokines, so as to lay a theoretical foundation for developing more efficient and stable AAV vectors.
References
- Earley LF, Conatser LM., et al. "Adeno-Associated Virus Serotype-Specific Inverted Terminal Repeat Sequence Role in Vector Transgene Expression." Hum Gene Ther. 2020 31(3-4):151-162 https://doi.org/10.1089/hum.2019.274
- Hanlon KS, Kleinstiver BP., et al. "High levels of AAV vector integration into CRISPR-induced DNA breaks." Nat Commun. 2019 10(1):4439 https://doi.org/10.1038/s41467-019-12449-2
- Hewitt FC, Samulski RJ. "Creating a novel origin of replication through modulating DNA-protein interfaces." PLoS One. 2010 5(1):e8850 https://doi.org/10.1371/journal.pone.0008850
- De Ravin SS, Reik A., et al. "Targeted gene addition in human CD34(+) hematopoietic cells for correction of X-linked chronic granulomatous disease." Nat Biotechnol. 2016 34(4):424-9 https://doi.org/10.1038/nbt.3513
- Chen HM, Resendes R., et al. "Molecular characterization of precise in vivo targeted gene integration in human cells using AAVHSC15." PLoS One. 2020 15(5):e0233373 https://doi.org/10.1371/journal.pone.0233373
- Mroske C, Rivera H., et al. "A capillary electrophoresis sequencing method for the identification of mutations in the inverted terminal repeats of adeno-associated virus." Hum Gene Ther Methods. 2012 23(2):128-36 https://doi.org/10.1089/hgtb.2011.231


